导图社区 《微习惯:简单到不可能失败的自我管理法则》
用一个简单到不可能失败的行动去培养一个习惯,行动只是手段,习惯本身才是目的。这是微习惯养成的指导手册,可以拿来就用,而且不需要付出太多的意志力和精力。
所谓SOP,是标准作业程序,指将某一事件的标准操作步骤和要求以统一的格式描述出来,用于指导和规范日常的工作。这里介绍了SOP(标准作业程序)的定义,作用和优化,自己我们为什么需要它。
这是一篇关于如何正确吵架的思维导图。只有学会正确的吵架方式,勇敢吵,坦诚吵,才有机会解决你们各自身上和亲密关系中存在的问题。
社区模板帮助中心,点此进入>>
论语孔子简单思维导图
《傅雷家书》思维导图
《童年》读书笔记
《茶馆》思维导图
《朝花夕拾》篇目思维导图
《昆虫记》思维导图
《安徒生童话》思维导图
《鲁滨逊漂流记》读书笔记
《这样读书就够了》读书笔记
妈妈必读:一张0-1岁孩子认知发展的精确时间表
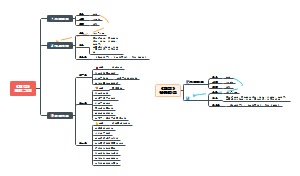
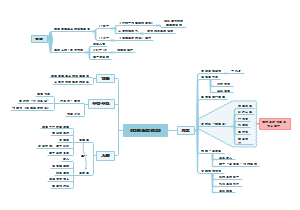
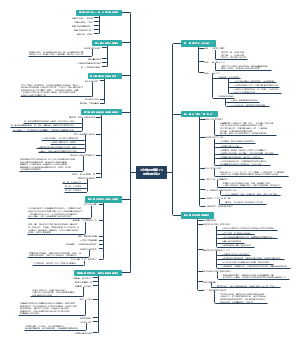
《微习惯》
是什么
目的:只为培养好习惯
方法:设定并完成简单到不可能失败的目标
例如1:一天做一个俯卧撑
例如2:每天写50个字
优点
完成目标的成就感
可能会进行“额外环节”的积极行为(例如设定一天做一个俯卧撑,结果做了五个)
惯性:即使没有额外完成,也会发展成为微习惯
影响因素
大脑中的神经通路是习惯的生物学原理
压力促进人更加依赖惯性
养成习惯需要多久?
误区:21天养成习惯
平均时间66天,按不同行为从18到154天不等
习惯没有开关键,养成习惯后抵触情绪会减弱
原理
大脑工作
意识部分——前额皮层:监督自发行为,发现能改进的地方就会介入
潜意识部分——基底神经节:自动,高效率,无需监督处理任务
驱动
动力
动力不可靠,行为的难易程度需要动力不同
不会每次都愿意激发动力
热情递减,动力逐渐衰退
意志力
误区
意志力取之不尽
意志力不能养成习惯
特点
意志力可以被强化
意志力可以通过计划执行
做决定会消耗意志力
策略
对抗意志力自我耗损
努力程度————微习惯需要非常少的实际努力
感知难度————微习惯的本质决定了它根本就不困难
消极情绪,指不愉快的体验————微习惯的完成是一件开心的事
主观疲劳————主观疲劳无法消除,但微习惯可以有效缓解
血糖水平————和微习惯相对独立,但微习惯会节约能量和意志力
对抗阻力
阻力出现的时间节点
行动前的阻力
继续行动时面对的阻力
阻力的类型
精神阻碍:你有锻炼的精力却不想锻炼————运用意志力抵抗
身体阻碍:因为很累所以身体不想锻炼————目标从易到难
独特性
微习惯能与现有习惯一较高下
微步骤+意志力是必胜组合:微步骤几乎不需要意志力,所以留给你更多的意志力
微习惯没有截止时间
微习惯每天的成功能够提升自我效能感
微习惯能够给予你自主权:实践轻松所以没有被计划控制
抽象和具体目标与微习惯相结合:具体、微小的目标是抽象目标的拆分
远离恐惧、怀疑、胆怯或犹豫
增强正念和意志力
步骤
第一步:选择适合你的微习惯和计划
设定计划
一周弹性计划
测试和评估一个长期计划
单一微计划
多项微计划
根据个人自控力而定
把习惯变成“小得不可思议的一小步”
第二步:挖掘每个微习惯的内在价值
微习惯来源于生活,认清其来源
反问自己“为什么”寻找核心因素
例如:我想每天写作。为什么?
因为我热爱写作。为什么?
因为表达想法、讲故事的方式是我最喜欢的,能通过写作和人们建立联系。为什么?
因为这些事情让我特别有活力、特别幸福。为什么?
因为写作是生活中我认为有价值并极为重视的事情。
第三步:明确习惯依据,将其纳入日程
计划依据
根据时间:规律、稳固但缺乏灵活变通———例如:每天3点到9点完成。
根据行为方式:更灵活但也更含糊————例如:每天午饭后、工作前完成。
选择自由度高的非具体习惯
自由度高:更自主、自由和灵活
非具体:不给意志力增加负担
第四步:建立回报机制,以奖励提升成就感
回报关联:例如每次完成任务都对自己竖个大拇子
回报策略:展望即将养成的习惯
回报对意志力重建作用
第五步:记录与追踪完成情况
大日历:记录每天的完成情况
数字化(软件)
IOS:Lift
安卓:Habit Streak Plan
电脑:Joe'sGoals
第六步:微量开始,超额完成
微量开始
不耗尽意志力
安全线:就算你只达到最低目标,依然能够形成习惯
超额完成
强化意志力
激发动力
当下就取得进步
第七步:服从计划安排,摆脱高期待值
误区:超额完成后,设定新的高于设定的目标
摆脱高期待
高期待带来负担和压力
把期待值放在坚持目标上,而非任务量
第八步:留意习惯养成的标志
没有抵触情绪
认同当下行为
行动时无需考虑
不再担心漏掉一天或过早放弃
常态化
它很无聊:好习惯不会让人兴奋,只是对你有好处而已
规则
1. 绝不要自欺欺人
如果完成不了,那就降低目标
不要提高期待值,反而要调低期待值
2. 满意每一个进步
3. 经常回报自己,尤其在完成微习惯之后
4. 保持头脑清醒并信任你选择的策略
5. 感到强烈抵触时,后退并缩小目标
6. 提醒自己这件事很轻松
7. 绝不要小看微步骤
8. 用多余精力超额完成任务,而不是制定更大目标