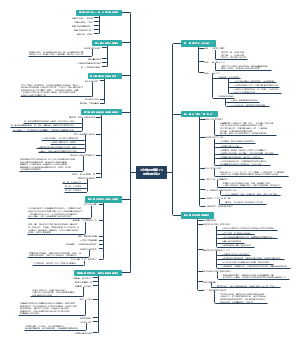
导图社区 地中海贫血
地中海贫血
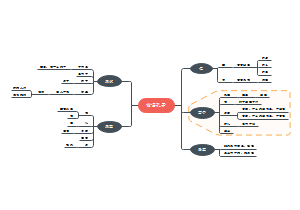
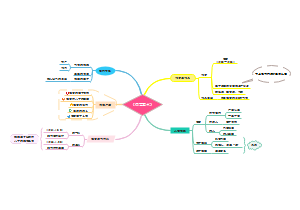
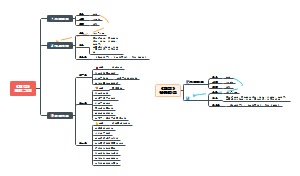
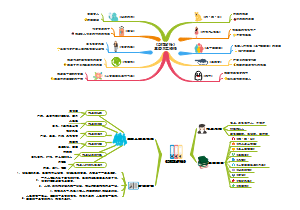
本图从地中海贫血的简介、病因基础、临床表现、诊断治疗以及预防详尽的介绍地贫这种遗传病的相关知识。地中海贫血是一种遗传性 的血液疾病。此病的特点 是红血球中一种称为血红 蛋白的物质制造不足。血 液藉著血红蛋白将氧气及营养送至全身各个 部位。如果氧气供应不充足,身体各部位的 器官及组织的运作便会受影响。
编辑于2021-07-11 18:55:58- 地中海贫血
- 心理现象研究
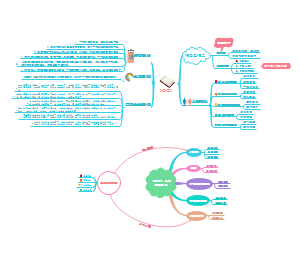
心理世界如同动态拼图,涵盖认知、情感、行为的复杂互动从感知觉异常到人格解体,从强迫行为到心境障碍,每个现象都揭示大脑与环境的博弈学习积累、文化影响塑造个性倾向,而情绪障碍、思维障碍则暴露系统脆弱性治疗需多管齐下认知行为疗法重整扭曲思维,药物调节生化失衡,社会支持修复人际裂痕无论是注意力的流动还是价值观的固化,心理活动始终徘徊于稳定与失衡的边缘,等待科学解码。
- 心理过程
探索心理奥秘:解码行为与情感的密码 心理过程涵盖认知、情绪情感与意志,构成我们对外界的反应基础个性心理特征如能力、气质、性格,塑造独特的行为模式与态度个性倾向性则决定行动方向这些因素交织影响知识经验奠定认知效率,情感基调左右情绪表现,而态度与风格赋予心理活动独特印记从内在情感到外在行为,心理机制无声支配着每一刻的选择与成长。
- 心理学流派与人物
心理学流派与关键发现速览 心理学发展脉络丰富,从冯特的构造主义到弗洛伊德的精神分析,再到马斯洛的人本主义,不同流派塑造了人类行为与心智的研究框架经典理论如韦伯定律揭示感知规律,耶克斯道德森定律解释动机效率的倒U关系安斯沃斯通过陌生情境实验划分依恋类型(65%安全型),而吉布森的视崖实验证明婴儿深度知觉社会心理学中,霍曼斯的交换理论强调互惠,勒温的场论提出行为是人与环境的函数这些基石理论至今影响临床、教育与社会互动研究。
地中海贫血
社区模板帮助中心,点此进入>>
- 心理现象研究
心理世界如同动态拼图,涵盖认知、情感、行为的复杂互动从感知觉异常到人格解体,从强迫行为到心境障碍,每个现象都揭示大脑与环境的博弈学习积累、文化影响塑造个性倾向,而情绪障碍、思维障碍则暴露系统脆弱性治疗需多管齐下认知行为疗法重整扭曲思维,药物调节生化失衡,社会支持修复人际裂痕无论是注意力的流动还是价值观的固化,心理活动始终徘徊于稳定与失衡的边缘,等待科学解码。
- 心理过程
探索心理奥秘:解码行为与情感的密码 心理过程涵盖认知、情绪情感与意志,构成我们对外界的反应基础个性心理特征如能力、气质、性格,塑造独特的行为模式与态度个性倾向性则决定行动方向这些因素交织影响知识经验奠定认知效率,情感基调左右情绪表现,而态度与风格赋予心理活动独特印记从内在情感到外在行为,心理机制无声支配着每一刻的选择与成长。
- 心理学流派与人物
心理学流派与关键发现速览 心理学发展脉络丰富,从冯特的构造主义到弗洛伊德的精神分析,再到马斯洛的人本主义,不同流派塑造了人类行为与心智的研究框架经典理论如韦伯定律揭示感知规律,耶克斯道德森定律解释动机效率的倒U关系安斯沃斯通过陌生情境实验划分依恋类型(65%安全型),而吉布森的视崖实验证明婴儿深度知觉社会心理学中,霍曼斯的交换理论强调互惠,勒温的场论提出行为是人与环境的函数这些基石理论至今影响临床、教育与社会互动研究。
- 相似推荐
- 大纲