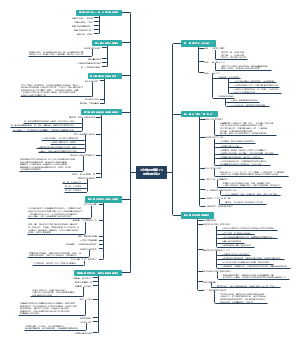
导图社区 地方各级人民代表大会各委员会(人大各专委)
地方各级人民代表大会各委员会(人大各专委)
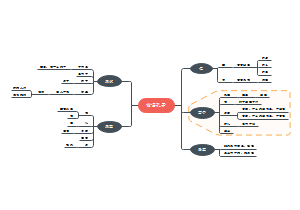
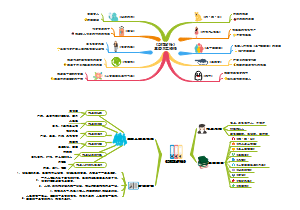
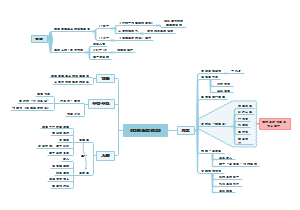
这是一个关于地方各级人民代表大会各委员会(人大各专委)的思维导图,概述了“特定问题调查委员会”的组成、任期、以及大会期间和闭会期间的不同职能。这有助于人们更好地理解特定问题调查委员会在人民代表大会制度中的角色和职责。
编辑于2024-05-28 09:46:07- 职责
- 人民代表大会制度
- 组成人员
他的近期作品
查看更多>>
- 地方各级人民代表大会各委员会(人大各专委)
这是一个关于地方各级人民代表大会各委员会(人大各专委)的思维导图,概述了“特定问题调查委员会”的组成、任期、以及大会期间和闭会期间的不同职能。这有助于人们更好地理解特定问题调查委员会在人民代表大会制度中的角色和职责。
- 地方各级人民代表大会各委员会(人大各专委)
组织法第二章第五节地方各级人民代表大会各委员会(人大各专委),大会期间,由主席团在代表中提名,大会通过;闭会期间,常委会可以任免个别副主任委员和部分委员,由主任会议提名,常委会会议通过。
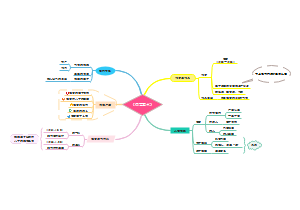
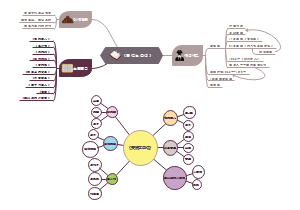
- 标题写作技巧——读《人大新闻消息写作技巧》
标题,是引读者来看文章的“酒旗、幌子、招牌”。在现在信息大爆炸的时代,标题决定稿件的命运。
地方各级人民代表大会各委员会(人大各专委)
社区模板帮助中心,点此进入>>
他的近期作品
查看更多>>
- 地方各级人民代表大会各委员会(人大各专委)
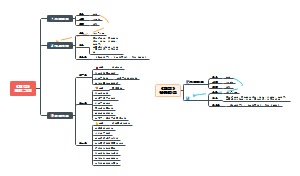
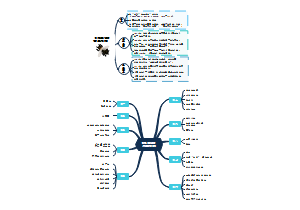
这是一个关于地方各级人民代表大会各委员会(人大各专委)的思维导图,概述了“特定问题调查委员会”的组成、任期、以及大会期间和闭会期间的不同职能。这有助于人们更好地理解特定问题调查委员会在人民代表大会制度中的角色和职责。
- 地方各级人民代表大会各委员会(人大各专委)
组织法第二章第五节地方各级人民代表大会各委员会(人大各专委),大会期间,由主席团在代表中提名,大会通过;闭会期间,常委会可以任免个别副主任委员和部分委员,由主任会议提名,常委会会议通过。
- 标题写作技巧——读《人大新闻消息写作技巧》
标题,是引读者来看文章的“酒旗、幌子、招牌”。在现在信息大爆炸的时代,标题决定稿件的命运。
- 相似推荐
- 大纲