导图社区 锂电池性能设计
- 727
- 59
- 3
- 举报
锂电池性能设计
锂电池性能设计,包括倍率、自放电、高温、低温、内阻、电压、容量、循环寿命等内容,有兴趣的可以看下。
编辑于2022-04-14 17:27:03- 锂电池
- 锂电池性能设计
- 相似推荐
- 大纲
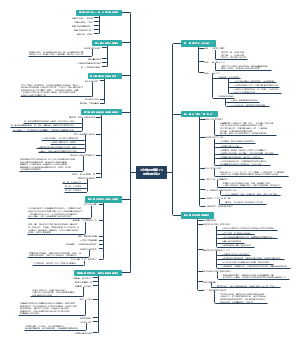

锂电池性能设计
倍率
正极
电导率
LiCoO2
10-3S·cm
LiMn2O4
10-4S·cm
LiFePO4
10-9~10-10S·cm
LiNixCoyMn1-x-yO2
10-4 S·cm
离子扩散系数 锂离子扩散系数大,锂离子在材料内部的移动更迅速, 嵌入和脱嵌的能力强。是影响电芯内阻的因素,也是影响功率特性的因素。
LiCoO2二维离子通道,层状结构;锂离子扩散系数较小
10-10~10-12
LiMn2O4三维离子通道;锂离子扩散系数较大
10-9~10-11
LiFePO4
10-13~10-16
随着含碳量升高,电池内阻逐渐减小,内阻越小,倍率性能越好。碳含量控制在 1~3 之间(碳含量增高,直接影响正极有效活性物质的量,导致材料克容量发挥,降低电池能量密度;②包覆碳一般比表面积远大于LFP材料本身比表面积,随着碳含量增加,LFP材料比表面积大幅增加,导致了在正极浆料配制过程中材料团聚,难以分散均匀,易出现“果冻型”、“颗粒型”等多种不良状况,影响电池良品率加工效率)
LiNixCoyMn1-x-yO2 二维离子通道,层状结构;锂离子扩散系数较小
10-11
采用量子化学第一性原理理论计算发现三元材料,存在两种类型的锂离子扩散通道ODH和TSH,在充电的初始阶段以ODH为主,随后以TSH为主。对于两类通道,Ni2+和Ni3+附近的锂离子扩散能垒最低,形成优势通道。同时Mn和Co等过渡金属元素会对结构、缺陷和锂层的层间距起到调制作用,从而间接影响锂离子的扩散
粒径
小粒径材料,锂离子由晶体结构迁移到电解液中的路径更短,提升离子传输速率,离子电导率更高,因而极化更小,直接提高倍率性能 (粒径较小,比表面积较大,与电解液接触面更大、副反应更多造成,使其首次库仑效率较低。)
粒径较小的三元材料的阳离子混排更为严重,这也会使其首次库仑效率较低。
比表面积
材料比表面积大,材料与电解液接触充分,增大锂离子扩散面积和通道,提高锂离子迁移速率,提高倍率性能(球的比表面最大,所制备球形颗粒对锂电池正极材料有利,颗粒越小,比表面积越大;但是比表面积越大,颗粒表面能升高,易团聚并与电解液反应,存在安全隐患)
LiCoO2 0.2~0.9m2/g
LiMn2O4 0.5~2.0m2/g
LiFePO4 ≤30m2/g
LiNixCoyMn1-x-yO2 0.3~0.8m2/g
负极
离子扩散系数
石墨化中间相沥青炭微球的球形片层结构利于锂离子从球的各个方向嵌入和脱出,减小了锂离子在固相中的扩散电阻,从而提高电极的高倍率性能。
粒度
减小颗粒粒径,有利于缩短 Li+ 脱嵌路径,在相同扩散速度下由于半径较小,在相同时间内锂离子更易嵌脱,提高电池倍率充放电性能 ;而对于粒径较大的炭微球而言,由于扩散路径较长, 锂离子不容易到达微粒的中心,这样就使微粒内部的一部分嵌锂位没有被活化,从而使容量降低。
比表面积
材料尺寸较小,比表面积较大,一方面可使电极的电流密度降低,减轻电极的极化作用;另一方面可提供更多的 Li+迁移通道,缩短迁移路径,降低扩散阻抗,因此倍率性能较好。
导电剂
电导率
电导率高则导电性强,电子和离子迁移速率快,降低活性物质之间、活性物质与正极基体/集流体的接触电阻,改善正极材料的电导率(离子和电子电导率),提升倍率性能。
比表面积
乙炔黑 55~70m2/g
Super-P 62m2/g
科琴黑 400~1000m2/g
350G 770m2/g
碳纤维VGCF 13~20m2/g
碳纳米管CNTs 400m2/g
KS 20m2/g
SFG 20m2/g
结构类型
颗粒状
线状
粒度
Super-P:30~40nm
颗粒较小,具有链式结构,能够将活性物质颗粒紧紧吸附在一起,这些支点可由导电炭黑 SP 充当
碳纤维VGCF:150nm
碳纳米管CNTs管径5nm
支点与节点之间还需要连接的导线,构筑成完整的网络,它们需要有极佳的导电性,较长的线状结构,ECP,VGCF 和 CNTs 是符合这一要求的备选材料(表面积很大,导电率高,难以分散的物质)
通过不同形状,不同导电能力的导电材料配合使用,构筑有节点,支架的完整导电网络,达到最好的电池性能
电解液
离子电导
粘度
粘度越小,电解液离子电导率越大;(粘度随锂盐浓度增大而增大,随阴离子半径增大而增大)
锂离子溶剂化半径
离子在电解液中的溶剂化半径越小,电解液的电导率就越大
锂盐浓度
锂盐浓度大,离子电导率大,但是锂盐浓度大会增加电解液粘度,反而会降低离子电导率
锂盐电导率高的电解液,自由离子数量多,且离子迁移速度快,在高倍率放电的情况下,可实现Li+的迅速补充与迁移,使放电过程得以顺利进行。
粘接剂
比表面积
粒度
绝电系数
隔膜
厚度、润湿
隔膜与电解液润湿性越好,电解液与其接触越充分,扩大隔膜与电解液的接触面,提高锂离子迁移速率。
基膜表面涂覆纳米级陶瓷颗粒,纳米级颗粒具有较大的比表面积增加隔膜对电解液的润湿性及保液能力
孔隙率
孔隙率更高,保液性更好,有利于 Li+ 在正负极之间迁移,减小极化,提高电池倍率充电性能。
采用高孔隙率的双面陶瓷涂层隔膜,可提升 Li+ 迁移速率,减小充放电过程中的极化作用,有利于提高电池倍率性能。
孔径
孔径太小会增加电阻,增加充放电过程中的极化作用,不利于提高电池倍率性能。(孔径太大容易使正负极接触或被枝晶刺穿短路,亚微米孔径的隔膜足以阻止电极颗粒直接通过)
集流体
厚度
电阻=电阻率×电阻长度/电阻截面积,可知更厚的铝箔会减小内阻。更厚的集流体,有更好的导电性和导热性,有效地改善电池倍率放电性能。(集流体越厚越好,但电池内部空间是确定的,更薄的集流体意味着更高的能量密度)
导电涂层
导电涂层有效减小了活性材料与集流体之间的接触电阻,减小电池放电过程中的极化,在更大倍率的放电和低温倍率放电时尤其重要。极化过大,瞬间的极化压降就会使电池电压低于截止电压以下,导致放电终止。
极耳
数量
增加极耳数量,可减小电池内阻,有利于增大电子传导和离子传导率,可减小欧姆产热,改善电池内部温度状态,提升倍率及循环性能。
位置
厚度
材质
隐形因子
SEI膜电导率
SEI由两层物质构成,内层主要成分是Li2CO3,而其外层主要成分是烷基碳酸锂如(CH2OCOLi)2等,负极表面SEI膜相当于锂离子扩散过程的一道门槛,影响锂离子嵌脱,高倍率下尤为明显
SEI膜厚度
SEI膜有一定的厚度,约100-120nm,其成分随电解液成分的不同而不同,一般由Li2O、LiF、LiCl、Li2CO3、LiCO2-R、醇盐和非导电聚合物组成,是多层结构,靠近电解液的一面是多孔的,靠近电极的一面是致密的,虽然锂离子可以穿透,但是SEI膜会造成负极表面部分扩散孔道的堵塞,不利于锂离子在负极材料的扩散
循环寿命
正极
结构稳定性
晶格结构
结构不稳定,导致在充放电循环中,晶格结构坍塌,晶粒形貌和尺寸变化,形成晶格缺陷,从而无法继续完成嵌锂脱锂,不可逆容量增加,循环寿命缩短。
化学稳定性
金属元素
正极中金属离子在电解液中的溶解,溶解的金属离子通过电解液迁移到负极,在负极表面以金属或者盐的形式沉积在负极表面,造成电极极化增大。
压实密度
真密度
真密度(g/cm3):
钴酸锂(5.1)>三元材料(4.8)>锰酸锂(4.2)>磷酸铁锂(3.6)
压实密度(g/cm3):
钴酸锂(4.1-4.3)>三元材料(3.4-3.7)>锰酸锂(2.9-3.2)>磷酸铁锂(2.2-2.3)
与压实密度成正比,材料的真密度对压实密度的影响是无法改变的
粒径分布
等径球在堆积时,球体和球体之间会有大量的空隙,若没有合适的小粒径来填补这些空隙,堆积密度就会很低,所以合适的粒度分布能提高材料的压实密度。粒度分布太窄或粒度分布太宽都会使材料压实密度降低,对于粒度分布的影响
包覆
压实过高,虽然可以提高电芯的能量密度,但会使电解液浸润不充分,充放电反应不完全,容量降低,循环性能下降。
颗粒形貌
目前商业化的钴酸锂是一次颗粒,单晶很大,三元材料则为细小单晶的二次团聚体,几百纳米的一次颗粒团聚成三元材料二次球,本身就有很多空隙;而制备成极片后,球和球之间也会有大量的空隙,以上原因使三元材料的压实密度进一步降低。
金属杂质
负极
SEI稳定性
SEI膜会消耗电池中有限的锂离子,若膜在循环过程中不断遭到破坏,在负极电解液界面将不断发生氧化反应形成新膜,会消耗体系中正极能提供的有限的锂离子,活性锂离子的减少会导致容量的衰减;SEI膜加厚,极化增加,内阻增加
结构稳定性
溶剂化锂离子的嵌层反应或者溶剂共嵌入反应,会直接破坏石墨层的结构,导致层状的石墨结构被粉化
层间距在嵌脱锂离子过程中,不断膨胀和收缩,导致负极体积变化大,结构被破坏,降低循环寿命
压实密度
真密度
粒径分布
包覆
颗粒形貌
金属杂质
导电剂
电导率
电导率差,增大活性物质之间、活性物质与正负极基体/集流体的接触电阻,循环性能差
比表面积
导电剂粒径大,比表面积小,不利于电解液吸附,与电解液接触面积小,不利于循环
粘结剂
化学稳定性
正负极的活性物质,是通过粘结剂固定在基体上面的,在长期使用过程中,由于粘结剂的失效以及电池受到机械振动等原因,正负极的活性物质不断脱落,进入电解质溶液,这导致能够参与电化学反应的活性物质不断减少,电池的循环寿命不断下降。
粘结剂的长期稳定性和电池良好的机械性能,将能够延缓电池循环寿命的下降速度。
电解液
保液量
虽然注液量充足但是老化时间不够或者正负极由于压实过高等原因造成的浸液不充分
电化学窗口
随着循环电芯内部电解液被消耗完毕。循环过程中,电解质受热分解和挥发,产生很多气体:
①形成安全隐患②有些气体对负极表面SEI膜产生破坏作用③长期累积下来,导致电解质总量减少,不能充分浸润正负极材料,充放电反应不完全,造成容量下降,影响循环性能。
杂质
水分
水会与电解质中的LiFP6发生化学反应,生产LiF和HF,HF进而又破坏SEI膜,生成更多的LiF,造成LiF沉积,不断的消耗活性的锂离子,造成电池循环寿命下降
HF
金属离子
电解液中铁、钠、铝、镍等金属离子杂质的氧化电位低于锂离子电池的正极电位,易在正极表面氧化,氧化物又在负极还原,不断消耗正负极活性物质,引起自放电,直接影响电池循环次数。
隔膜
吸液系数
孔隙率
润湿角
涂层
勃姆石
三氧化二铝
PVDF
膜结构
三层隔膜与单层隔膜相比,单层隔膜由于通常厚度较薄,离子迁移通道较短,极化现象有一定消弱,电池的低温电压平台相对较高。同理,采用薄隔膜或者大孔径隔膜的电池循环也表现相对较好。
隔膜吸液性能差,浸润性在反复充电过程中变差,导致隔膜干涸,电池的欧姆内阻增大,导致充放电通道堵塞,充放电不完全,电池容量无法回复到初始状态,大大降低了电池的容量和循环寿命。
集流体
耐腐蚀性
结构强度
隐性因素
SEI膜稳定性
正极与电解液副反应
负极与电解液副反应
电解液保有量
自放电
容量
正极
理论克容量
C=26.8n/M(AH/g) n为mol活性物质反应转移的电子的电量 M为活性物质的摩尔质量
LiNiO2 276mAh/g
LiCoO2 274mAh/g
LiMn2O4 148mAh/g
LiFePO4 170mAh/g
LiNixCoyMn1-x-yO2 270mAh/g
实际嵌脱锂系数 脱嵌前后,能隙变化较小的体系 对于锂离子保持前后的容量非常 有利。
LiNiO2
LiNiO2与Li CoO2一样,充放电过程中也发生从三方晶系到单斜晶系的可逆相变。当Li1-xNiO2中x≤0.5时,时,Ni2+离子较Co2+离子更易在有机电解质中发生还原。在循环过程中结构的完整性还能得到保持。若x>0.5锂氧化物在充放电循环过程中涉及斜方六面体及单斜晶的变化,LiyNiO2通常在0.3<y<0.9范围内循环。
脱嵌系数>0.60~0.62;实际容量>165.6~171.1mAh
LiCoO2
Li CoO2实际比容量只有理论比容量的40%~50%,且反复充放电过程中,活性物质结构在多次收缩膨胀后发生改变,导致LiCoO2松动脱落,造成内阻增大,容量减小。造成这种现象根本原因在于LiCoO2是Li+嵌人式化合物,充电时若过多的Li+(一半以上)从Li CoO2中脱出,LiCoO2会发生晶型改变而不再具有嵌入和脱出Li+的功能。充电4.5V以上时,Li+脱出量超过了50%使得LCO结构出现坍塌,可逆容量迅速下降。锂钴氧化物在完全充电状态下为六方晶体,理论容量的50%放电后生成新相单斜晶体,所以LiyCoO2通常在0.5<y<1.0范围内循环。
脱嵌系数>0.56~0.58;实际容量>153.4~158.9mAh
LiMn2O4
锂锰氧化物在充放电过程中存在2种不同结构变化:一是化学计量不变的情况下发生的相变化;二是充放电过程中锂嵌入和脱嵌量改变时例如锂锰氧化物完全充电发生的相变。例如锂锰氧化物完全充电脱锂形成γ-MnO2,可能在化学计量不变的情况下发生相变化,经ε-MnO2转变成没有活性的β-MnO2;当锂锰氧化物深度放电嵌入过分的锂时,原来的立方晶系尖晶石发生晶格扭曲,转变成四方晶系的尖晶石,即发生Jahn-Teller扭曲,晶胞中Z轴伸长15%,X和Y轴收缩6%,这样多次循环后,正极材料变会粉化,LiMn2O4在深循环下易出现容量衰减。
脱嵌系数>0.79~0.8;实际容量>116.9~118.4mAh
LiFePO4
脱嵌系数>0.79~0.8;实际容量>134.3~136mAh
两种 Li+脱嵌模型:径向模型和马赛克模型。在 Li+的脱嵌过程中,晶体内部 b 轴的形变引起晶粒的破裂,破坏了颗粒与导电添加剂和集流体之间的电接触,从而引起了电极的极化。
LiNixCoyMn1-x-yO2
由于Li+(0.76 Å)与Ni2+(0.69 Å)有相近的离子半径,富镍材料较易出现Ni2+向Li+空穴迁移的情况,导致产生结构的无序性;体积的反复变化导致活性材料产生裂纹及孔隙,随着循环的进行,材料结构逐渐由菱方结构转变成尖晶石相,在循环初期结构的激烈变化导致容量及电压的快速衰退。
脱嵌系数>0.65;实际容量>180mAh
压实密度
真密度 材料真密度越大同等条件 下的压实密度也越大容量越大
LiNiO2 g/cm3
LiCoO2 5.1g/cm3
实际值4.1~4.3g/cm3
LiMn2O4 4.2g/cm3
实际值2.9~3.2g/cm3
LiFePO4 3.6g/cm3
实际值2.2~2.3g/cm3
LiNixCoyMn1-x-yO2 4.8g/cm3
实际值3.4~3.7g/cm3
颗粒形貌 提高压实密度的途径:大单晶 一次颗粒比二次颗粒压实大 一次晶粒大材料结构稳定。
钴酸锂是一次颗粒,单晶很大,不存在二次颗粒内部间隙的影响,压实密度较大。 自有长成警惕表面光滑,与导电剂接触紧密。高温下长成的晶格缺陷少,锂离子传输 通畅;此外把粒径做小也有助于倍率提升。
子主题
锰酸锂是单晶颗粒中空隙小,压实密度大;球形形貌有利于颗粒堆积,压实密度降低;不规则形状,颗粒堆积内部空隙多,压实密度小。
磷酸铁锂由于材料的纳米化,限制了压实的进一步提高
三元材料则为细小单晶的二次团聚体,几百纳米的一次颗粒团聚成三元材料二次球,本身就有很多空隙;而制备成极片后,球和球之间也会有大量的空隙,以上原因使三元材料的压实密度进一步降低
子主题
粒径分布
LiNiO2
D10≥1.0μm,5.0≤D50≤10.0μm,D90<30.0μm
LiCoO2
D10≥1.0μm,5.0≤D50≤15.0μm,D90<30.0μm
LiMn2O4
D10≥2.0μm,4.0≤D50≤30.0μm,D90<50.0μm
LiFePO4
D10≥0.1μm,2.0≤D50≤10.0μm,D90<15.0μm
LiNixCoyMn1-x-yO2
D10≥2.0μm,5.0≤D50≤15.0μm,D90<30.0μm
粒度分布太窄或者粒度分布太宽都会使材料压实密度降低,细粉过多也影响压实。
保有重量
电池内部腾出更多的空间容纳更多的正极材料
负极
克容量
n/M比
天然石墨 372mAh/g
软体、硬碳、人造石墨 372mAh/g
硅碳 Li4Si 4211mAh/g(难以做到纯用)
压实密度
真密度
天然石墨 ≥2.22g/cm3
压实1.5~1.8g/cm3
人造石墨 ≥2.24g/cm3
压实1.5~1.8g/cm3
软碳 ≥1.7g/cm3
压实1.5~1.6g/cm3
硬碳 1.5~1.7g/cm3
压实1.3~1.5g/cm3
硅碳 ≥2.2g/cm3
不能单一材料使用没有参考压实
粒径分布
天然石墨
D10:11.0±2.0μm D50:18.0±2.0μm D90:30.0±3.0 μm Dmax≤60.0μm
人造石墨
D10:8.0±2.0μm D50:18.0±2.0μm D90:35.0±3.0 μm Dmax≤60.0μm
软碳、硬碳
等径球堆积时,球体间会有大量空隙,若没有合适的小粒径填补这些空隙,堆积密度就会很低,合适的粒度分布能提高材料的压实密度。
硅碳
纳米化
粒度分布太窄或粒度分布太宽都会使材料压实密度降低
研磨方式: 研磨频率高,粒度小,粒径分布宽
颗粒形貌
天然石墨
天然石墨大小颗粒不一,粒径分布广,一般呈鳞片状或板状,集合体呈致密块状、土状或球状,工业上将天然石墨分为致密结晶状石墨、鳞片石墨和隐晶质石墨三类。
人造石墨
形貌以及粒径分布上较一致
软碳、硬碳、硅碳
球形;球形石墨颗粒之间的接触明显不如不规则石墨颗粒的接触,因而阻抗也会大一些,这对材料的设计而言是一个方向,对颗粒大小的匹配以及保证颗粒之间的面接触,增大接触面积,降低接触阻抗,从而达到降低极化的目的。
实际嵌脱锂系数
石墨化度
循环后碳材料虽保持石墨的形貌结构,但其(002)晶面半高宽变大,导致c 轴方向晶粒尺寸变小,晶体结构的改变导致碳材料出现裂纹,进而破坏负极表面SEI 膜并促进SEI 膜修复,SEI 膜过度生长消耗活性锂,造成了电池不可逆容量衰减。(d002 呈现增加趋势,石墨化程度呈现减小趋势。石墨电极材料的晶粒尺寸Lc呈现降低趋势,La 没有出现明显变化规律。其中Lc是d002 与晶粒内石墨片层数的乘积,由此可以得到石墨片层数呈现减小趋势,这样的结构变化宏观上表现为石墨材料的脱落,最终导致锂电池容量衰减。
较高的石墨化度,储锂空间大,容量较大
体积膨胀
①硅材料在脱嵌锂过程中反复膨胀收缩,致使负极材料粉化、脱落,并最终导致负极材料失去电接触而使电池彻底失效;② 硅材料表面 SEI 膜的持续生长,会一直不可逆地消耗电池中有限的电解液和来自正极的锂,最终导致电池容量的迅速衰减
碳包覆纳米硅(nano-Si@C);氧化亚硅碳复合材料(SiO@C);硅纳米线(Si nannowire/SS);变氧型氧化亚硅碳复合材料((SixO@C);无定型硅合金((SiM)
cell balance
通过精确控制正负极的克容量发挥,把负极的余量尽可能缩小
提高涂布过程的公差控制能力
隔膜
厚度
PP
9μm;12μm;14μm;16μm;20μm
PE
5μm;7μm;9μm;11μm;14μm;16μm
PP/PE/PP
12μm;14μm;16μm;20μm
无机物涂层
1μm,2μm,单面,双面,水性,油性
无纺布
量产厚度:3~15μm
在保证机械强度情况下,在同样大小的电池中,隔膜越薄,越省空间,能卷绕的层数就越多,相应容量会增大;较厚的隔膜,穿刺强度会高,安全性会高。
面密度
原材料密度
PP;PE;多层结构;密度在8~12g/cm3
无机物涂层:20g/cm3
陶瓷(Al2O3、SiO2、ZrO2、MgO、黏土)和粘结剂(间芳香聚酰胺、SBR、PVDF、PEO、CMC)混合制备浆料涂覆在聚烯烃、无纺布隔膜上
无纺布: 20g/cm3
无纺布隔膜基材通常选用聚对苯二甲酸乙二醇酯(PET,熔点高达225℃)和聚酰亚胺(PI,熔点超过500℃)
孔隙率 保证机械强度下,尽可能增加隔膜孔隙率
PP;PE;多层结构;孔隙率在35~55%
无纺布孔隙率:30~75%
隔膜孔隙率越大,同等厚度的隔膜质量越小,在同样质量的电池中,隔膜所占质量越小,相应容量增加;隔膜孔隙率太大会使隔膜的机械强度变差;孔隙率太小,电池内阻增大。
导电剂
电导率
乙炔黑
Super-P
10S/cm
科琴黑
350G
碳纤维VGCF
1000S/cm
碳纳米管CNTs
297S/cm
KS
SFG
1000S/cm
电导率高的导电剂使得电池在大电流充放电时保持高容量和高循环效率;导电剂导电性顺序;SP系列<C系列<导电石墨(KS-6、SFG-6)<350G<科琴黑系列(EC300J<EC600JD<ECP<ECP600JD)<VGCF<CNTs
比表面积
乙炔黑 黑类型,介于Super-P和KS-6间,导电性较低,体积蓬松,对材料压实影响较大
粒度 30~45nm 比表面积55~70m2/g
Super-P 小颗粒导电炭黑;常用导电剂,价格便宜实用
粒度30~40nm 比表面积 62m2/g
科琴黑 超导炭黑;高纯度,具有独特的支链结构和优越的导电性能
粒度30~50nm 比表面积 400~1000m2/g
350G
粒度40nm 比表面积770m2/g
碳纤维VGCF 气相生长碳纤维;用于大功率大倍率电池,分散困难
粒度150nm 比表面积13~20m2/g
碳纳米管CNTs 直径在5nm左右,长度10~20μm,不仅在导电网络中充当导线作用,同时还具有双电层效应,发挥超级电容器的高倍率特性,其良好的导热性有利于电池充放电时的散热,减少电池的极化,提高电池的高低温性能
管径5nm 比表面积400m2/g
KS 大颗粒石墨粉;性能优于Super-P,价格较贵,为高容量电池使用,可与Super-P混用
粒径5~6μm 比表面积20m2/g
SFG
粒径5~7μm 比表面积20m2/g
选择比表面积较大的导电剂,导电剂与活性物质接触充分,导电性好,所需导电剂含量也相对较少
密度
导电剂的密度都比较小,在电池设计中影响度很低
粘接剂
粘度
PVDF
粉末;使用粘度5000~10000Pa.S
CMC
粉末;使用粘度800~1200mPa·S
SBR
12mPa·S
LA
7300~13000mPa·S
比表面积
粒度
粘结剂粒度小,比表面积大,与活性物质和集流体接触面积大,充放电过程中保证良好的粘结作用,有利于电池容量发挥;粒度太小,分散性差,易团聚
SBR 150~200nm
LA D50<1.2μm
密度
集流体
厚度
铜箔
6μm;8μm;10μm
铝箔
9μm;11μm;14μm
密度
铜箔 8.92g/cm3
铝箔2.71g/cm3
隐性因素
SEI于首次充放电间完成,形成伴随部分锂离子的消耗,锂离子被消耗造成的就是电池不可逆容量的增加,就降低了电极材料的充放电效率,并且SEI膜并不是稳定不变的,会在循环过程中不断破裂,露出来新的碳表面再与电解质反应形成新的SEI膜,这样会不断造成锂离子和电解质的持续损耗,导致电池的容量下降。
PC溶液中,形成的SEI膜不能完全覆盖表面,电解液很容易在石墨表面反应,产生不可逆容量。在纯EC做溶剂时,生成的SEI膜主要成分是(CH2OCOOLi)2,而加入DEC或DMC后,形成的SEI膜的主要成分分别为C2H5COOLi和Li2CO3。显然,后二者形成的SEI膜更稳定。在EC/DEC和EC/DMC的混合体系中,EC是生成SEI膜的主要来源,只有EC发生了分解,DEC和DMC的主要作用是提高溶液的电导率和可溶性,而不在于参与SEI膜的形成。
在选择合适电解液的基础上通过加入合适添加剂,能够形成更稳定的SEI膜,提高电极表层分子膜的稳定性,减少溶剂分子的共嵌入
添加剂名称 作用体系 最佳用量 改进效果
PS PC ~5% 首效可达90%
VC EC+DMC ~2% 容量/寿命明显提高
苯甲醚 EC+DEC ~1.6% 首效可达90%
ES PC 3~5% 首效可达92.9%
正极材料结构本身会发生不可逆的变化,比如原子重排等,造成一些储锂位置不可逆,也会造成首次库伦效率低,这样即使外界有剩余的锂离子也不能补充损失的容量。
电极表面存在其它杂质,在较低的充电电压下,这部分物质被氧化,氧化还原反应发生;但这部分反应只发生在充电过程,而非可逆的,故在放电过程中无法发生。
充电后期电池微短路,未发生氧化还原反应,也即无电极反应及Li+的迁移,但是仍有电子(也即电流)经过测试仪器,电池的表观容量持续增加
电压
正极
本征电极电势
LiCoO2
电压窗口(V):3.0~4.5
LiMn2O4
电压窗口(V):3.0~4.3
LiFePO4
电压窗口(V):2.7~3.65
LiNixCoyMn1-x-yO2
电压窗口(V):2.5~4.6
电极电势与电解液电化学窗口最高占据轨道HOMO匹配(氧化电位),HOMO轨道能量越低,抗氧化性越强,越不易与电解液反应。正极电极电势高于电解液电化学窗口氧化电位,正极与电解液反应,电解液分解,放出热量。
有利趋势
①锂离子脱、嵌过程中正极的氧化还原电位应该处于电解质的电压窗口内,防止电解质发生氧化或还原反应。
②电极电位尽量与电解液的电化学窗口最高占据轨道HOMO完美匹配
E+和E-分别表示正极、负极的费米能级,EL、EH分别表示最低未占用分子轨和最高占用分子轨道能量,Eg=EL-EH,Eg为热力学稳定的电解质电压窗口,当E+>EL,E->EH,SEI膜保持稳定。
负极
本征电极电势
①负极本征电极电势须低于电解质的LUMO。
负极本征轨道能量和电解质LUMO,HOMO的配置关系影响其电压特性
电解液
电化学窗口
最高占据轨道HOMO能量
EC -11.905
PC -11.906
DMC -11.822
DEC -11.455
EMC -11.542
最低空轨道LUMO能量
EC 1.242
PC 1.314
DMC 1.138
DEC 1.125
EMC 1.13
有利趋势
①使用拥有更宽的电化学窗口(尤其是LUMO更高)的电解液,在电解质里添加一些阻燃材料,将混合的离子液体和有机液体电解质改性成为不易燃的电解液(与此同时离子传导率σLi也不会降低太多)等手段也可以有效地提高安全性
②设计采用LUMO能量低于其他溶剂的添加剂。如在电解液中加入GCL+AL31,理论上GCL 可以优先还原分解,形成覆盖在电极表面的SEI膜,保护SEI膜在高电压平台的稳定性。
隐性因素
平台电压
LiCoO2 3.7V
LiMn2O4 3.8V
LiFePO4 3.4V
LiNixCoyMn1-x-yO2 3.6~3.7V
放电过程
典型恒流放电测试的电压曲线可以分3个阶段:
1)电池在初始阶段端电压快速下降,放电倍率越大,电压下降的越快;
2)电池电压进入一个缓慢变化的阶段,这段时间称为电池的平台区,放电倍率越小,平台区持续的时间越长,平台电压越高,电压下降越缓慢。
3)在电池电量接近放完时,电池负载电压开始急剧下降直至达到放电截止电压。
其他影响
①同一个电池,在同等剩余容量的情况下,电压值因放电电流的大小而变化。
放电电流越大,电压越低。在没有电流的情况下电压最高。
②环境温度对电池电压的影响,温度越低,同等容量电池电压越低。
③循环对电池放电平台的影响,随着循环的进行,锂离子电池的放电平台趋于恶化。放电平台降低。所以相同电压所代表的容量也相应变化了。
④不同厂家,不同容量的锂离子电池,其放电的平台略有差异。
⑤不同类型的电极材料的锂离子电池,放电平台有较大差异。
内阻
直流内阻(欧姆内阻+化学极化内阻)
隔膜
macmullin值
材料电阻
隔膜越薄内阻越小,从而实现大功率充放电。
吸液系数
接触角
为了保证电池的内阻不是太大,要求隔膜是能够被电池所用电解液完全浸润。较好的浸润性有利于提高隔膜与电解液的亲和性,扩大隔膜与电解液的接触面,从而增加离子导电性,提高电池的充放电性能和容量。浸润性可通过测定其吸液率和持液率来衡量。
孔隙率
由于隔膜含有大量微孔,随着微孔数量减少,隔膜面密度的增大,孔隙率、透气率会降低,内阻增大。不同厚度、不同工艺的隔膜,其面密度无可比性。但不同种隔膜之间的孔隙率的绝对值无法直接比较。
涂层
在电芯拆解之后经常会发现隔膜上沾着厚厚一层褐色物质,里面包括石墨负极及其反应副产物,会造成隔膜孔堵塞,降低电池使用寿命。
孔径/曲折度/厚度
一定体积的气体,在一定压力条件下通过一定面积的隔膜所需要的时间。与隔膜装配的电池的内阻成正比,即该数值越大内阻越大。不过单纯比较两种不同隔膜的Gurley数是没有意义的,因为它们的微观结构可能完全不一样,但是同一种隔膜的Gurley数的大小可以很好的反应内阻的大小。
粘结剂
电阻率
分子量
粘结剂一般都是高分子材料,绝缘性能较强,若含量过高,会降低导电性
分子构型
电解液
离子电导率
粒度锂离子溶剂化半径
锂盐浓度
电解液也需要较低的浓度,浓度过高同样不利于其流动浸润。
正极
电导率
电子电导率(20℃):
LCO材料最低仅为5x10-8S/cm,
LFP10-9cm/s
NCM111材料电子电导率可达2.2x10-6S/cm;
随着镍含量的进一步提高,三元材料的电子电导率也明显提高,NCM8111材料更是达到4.1x10-3S/cm。
离子电导率(20℃):
LCO仅为2.3x10-7S/cm;
NCM111为3.2x10-6S/cm;
NCM532为1.7x10-3S/cm;
NCM622为3.4x10-3S/cm;
NCM811为6.3x10-3S/cm
锂离子扩散系数
晶格结构
锂离子嵌入和脱嵌的难易程度,决定了材料内阻的大小,是浓差极化电阻的一部分。
压实密度
活物质密度太小时,颗粒间隙大,不利于电子传导。
压实密度过大,孔隙率小,电解液不易浸润,会提高离子阻抗。
正极片的辊压厚度影响着活性物质与集流体之间的接触电阻
相同辊压厚度时,阻抗的大小与面积成正比,面积为1时阻抗为a,面积为2时阻抗是2a
金属杂质
负极
材料内阻
SEI膜
如果首次充放电时在活物质表面形成的SEI膜过厚,也会提高离子阻抗。
锂离子扩散系数
晶格结构
锂离子嵌入和脱嵌的难易程度,决定了材料内阻的大小,是浓差极化电阻的一部分。
导电剂
电导率
集流体
面阻抗
材料电阻
厚度
表面氧化物
交流阻抗
隔膜
电阻
材料电阻
吸液系数
接触角
孔隙率
图层
厚度
曲折度
孔径
正极
电导率
负极
电导率
粘结剂
电导率
分子构型
分子量
极耳
数量
铜铝箔与极耳焊接不良也会影响电子阻抗。
位置
厚度
材质
集流体
电阻率
材料电阻
厚度
表面氧化物
面阻抗
低温
正极
离子扩散系数
锂离子扩散系数与温度的平方成正比,低温条件下,锂离子扩散系数降低,电池的电荷迁移阻抗增大,导致锂离子在正极中的扩散速率降低
结构稳定性
晶格结构
正极材料的三维结构制约着锂离子的扩散速率,低温下影响尤其明显,材料晶格在低温下稳定性差,晶格转变或坍塌,放电容量损失大。不同正极材料具有不同的三维结构,低温性能:LMO>NCM三元>LFP。
1.采用导电性优异的材料对活性物质本体进行表面包覆的方法提升正极材料界面的电导率,降低界面阻抗,同时减少正极材料和电解液的副反应,稳定材料结构;
2.通过Mn、Al、Cr、Mg、F 等元素对材料本体进行体相掺杂,增加材料的层间距来提高 Li+在本体中的扩散速率,降低 Li+的扩散阻抗,进而提升电池的低温性能;
3.降低材料粒径,缩短 Li+迁移路径. 该方法会增大材料的比表面积从而与电解液的副反应增多
负极
离子扩散系数
低温环境下锂离子在石墨负极中的扩散速率降低。负极析锂严重,并且析出的金属锂与电解液反应,其产物沉积导致固态电解质界面(SEI)厚度增加。负极在低温时的嵌锂阻抗明显大于正极脱锂阻抗,虽然锂离子可以在低温下相对快速的从正极脱嵌,但是却无法及时嵌入到负极当中,从而引发析锂。
电解液的低温电导率对锂离子电池低温性能的影响并没有想象中的大,负极的SEI膜的成分和结构对于电池的低温性能的影响要重要的多。SEI为锂离子电池在低温环境下的主要阻抗。
结构稳定性
粒径越大, 锂离子扩散路径越长,扩散阻抗越大,导致浓差极化增大,低温性能变差。
适当减小负极材料颗粒尺寸,可以有效缩短锂离子在石墨层间的迁移距离,降低扩散阻抗,增加电解液浸润面积,进而改善电池的低温性能.
石墨负极的层间距小, 低温下锂离子在石墨层间的扩散速率降低,导致极化增大
在石墨制备过程中引入 B、N、S、K等元素可以对石墨进行结构改性,增加石墨的层间距提高其脱/嵌锂能力
电解液
离子电导率
粘度
由于电解液混合溶剂中存在高熔点溶剂,低温环境下,电解液的黏度增大,当温度过低时甚至部分凝固,导致锂离子在电解液中迁移阻力大,传输速率降低,锂离子电池的导电率下降。
通过调整电解液溶剂的配比,就可以显著提升电解液的低温性能。环状的碳酸酯类溶剂会降低电解液的低温性能,而直链状溶剂则能够提升电解液的低温性能。
锂盐浓度
低温下,锂盐溶解度降低;
链状碳酸酯对锂盐的溶解度小,在低温下,电解液会发生固液相分离
1) 溶剂成分优化。采用熔点较低的 PC 溶剂部分取代EC 溶剂降低电解液熔点,以及通过低熔点、低黏度的共溶剂降低 EC 基电解液粘度并提高电解液低温电导率;2) 锂盐优化。加入低温性能较好的锂盐,改善电池低温性能;3) 低温添加剂。
加入效果优于 EC 的成膜添加剂降低低温下电极界面电荷传递阻抗,或加入锂盐沉积改善剂,防止低温下锂支晶生长,改善电池低温循环稳定性
锂盐溶解度
电解液的低温共溶剂。如甲酸甲酯(MF,熔点一99℃)、乙酸甲酯(MA,熔点~98℃)、乙酸乙酯(EA,熔点一83℃)、丙酸乙酯(EP,熔点一73℃)、丁酸甲酯(MB,熔点一84-93℃)和丁酸乙酯(EB,熔点-93℃)等以及成膜添加剂等
溶剂介电常数ε越大、离解作用越强,溶质锂盐的电离程度越大,则离子电导率越高。低温条件下,电解液离子电导率降低,锂离子扩散阻力增加,相反,电解液电导率越大,离子传导速率越快,低温性能越好。
①电解液改性;②采用高介电常数的环状碳酸酯和低黏度的链状碳酸酯的混合溶剂;
导电剂
电导率
粘结剂
粘接性
点状粘结剂会有极片粘结剂失效的情况,线状粘结剂不存在结构失效的问题
隔膜
吸液系数
低温下,隔膜与电解液相容性差,导致锂离子迁移阻力增大
隐性因素
SEI膜
好的SEI膜应该含有更多的LiF,从而减少Li+在SEI膜中的扩散阻抗。较多的链状溶剂,例如EMC和DMC,较少的环状溶剂,例如EC能够有效的提高锂离子电池的低温性能,但是为了形成更加稳定的SEI膜,我们还是需要添加少量的EC和PC。
隔膜与电解液相容性
析锂
层间距
正极负极和隔膜之间的间隙可以通过采用PVDF涂层来增强
高温
正极
热稳定性
热分解温度
LiNiO2
200℃左右
LiNiO2的热稳定性差,其热分解温度较低,,Ni由三价变为二价,且随着锂含量的减少,分解温度逐渐降低。由于充电后高氧化态的Ni4+不稳定,氧化性强,不仅氧化分解电解质,腐蚀集流体,放出热量和气体,而且自身不稳定,在一定温度下容易放热分解并析出O2。
LiCoO2
200℃左右
温度超过200℃,钴酸锂发生热分解,释放氧气,有机溶剂燃烧。
LiMn2O4
180℃
适当地提高正极材料尖晶石LiMn2O4的颗粒尺寸能在一定程度上减小在电解液中的溶解。
①高温循环下容量衰减的主要原因有:锰的溶解;电解液氧化;Jahn-Teller效应。
②充电态LiMn2O4热分解中有两个主要放热过程,通过计算得到其热分解活化能和指前因子,充电态LiMn2O4的在不同温度下的热分解反应速率常数不同。
LiFePO4
900℃
①磷酸铁锂开始发生化学键断裂的温度差不多在900℃左右。
②高温会使LiFePO4电极材料中的铁元素加速溶解于LiPF6酸性电解液中, 分解后的亚铁离子通过电解液传导, 沉积于电池负极石墨的表面,影响SEI膜生长和传质过程。
①研究可抑制铁元素溶解的电解液(如LiBOB电解液)、阳极材料(如Li4Ti5O12),以减少铁元素的溶解沉积。
LiNixCoyMn1-x-yO2
195℃
在195℃处出现升温热流曲线峰值, 该分解过程可能为正极表面亚稳定层物质的分解。
结构稳定性
晶格转变温度
LiMn2O4
780~1060℃下,LiMn2O4 通过失去氧和氧化锂, 产生新的物相。
相对于层状结构的LiCoO2 和LiNiO2,尖晶石结构的LiMn2O4具有最好的热稳定性。
780~1060℃下,LiMn2O4 通过失去氧和氧化锂, 产生新的物相。
持续高温或酸性条件下Mn会发生溶解,溶解的Mn2+被2Li+所取代,其中一半Li+进入八面体16d 位,另一半进入16c位。
LiFePO4
20℃~60℃对磷酸铁锂影响不大,在一定范围高温条件下,离子的扩散速度加快,有利于材料电化学性能的提高,放电容量增加;
55℃比室温内阻增加率大,这主要是由于在高温下,电极表面的固体电解质相界面膜(SEI膜)会分解、重组并增厚,导致活性锂离子的消耗,内阻增大,化学稳定性及容量降低。
LiNixCoyMn1-x-yO2
60℃环境下,正极材料表面非电化学活性的岩盐相厚度增加,并且晶粒内的局部范围有尖晶石相出现,可导致高镍三元材料可逆容量的衰减。
60℃存储过程中Ni和Mn元素从正极溶出,两种元素扩散到负极后发生还原,以金属Ni和金属Mn的形式沉积在负极上。
通过掺杂稳定三元材料的层状结构,在一定程度上抑制存储过程中向尖晶石和岩盐相的转变。
包覆处理减少电极/电解液界面副反应发生,稳定材料表面结构,抑制过渡金属元素的溶出。
保证充放电过程中,锂离子在固相中通畅、快速迁移。高温条件下,正极材料晶格转变,金属与氧间结合力不强,发生析氧反应,氧或正极材料与电解质等反应,放出热量,导致热失控。
化学稳定性
副反应温度
Li0.5CoO2 /电解液(LiPF6/EC/DEC) 130℃
Li0.15Mn2O4/电解液(LiClO4/EC+DMC) 220℃
LiFePO4/电解液(LiPF6/EC/DEC) 190℃
Li0.17FePO4/电解液(LiClO4/EC+DMC) 230℃
LiNixCoyMn1-x-yO2/电解液(LiPF6/EC+DEC+EMC)290℃
包覆处理,减少电极/电解液界面在高温时的副反应发生,稳定材料表面结构,抑制过渡金属元素的溶出
负极
热稳定性
热分解温度
①负极表面的膜由稳定态物质(如Li2CO3)和亚稳定态物质如(CH2O-CO2Li)2等两部分构成,当电池温度升高时,膜发生放热分解,分解反应为膜中的亚稳定态物质分解生成稳定态物质。
结构稳定性
体积膨胀率
天然石墨及硅碳的膨胀率大
电解液
热稳定性
热分解温度
LiPF6/EC+DMC(1:1) 250℃~280℃
LiPF6/PC+DC+EMC(1:1:3)215℃
锂离子电池的电解液中各组分的种类和比例是影响电解液热分解反应的主要因素。
在85℃,电解液中存在以下平衡:LiPF6→LiF+PF5,分解产物PF5和EC优先发生发应,生成可溶性单聚物、二聚物、齐聚物,同时还伴有不溶性的酸盐和磷酸醚类物质。
随着电解液中LiPF6浓度的降低,EC含量越低,DMC含量越高,电解液的分解温度都有明显的提高,稳定性更高。
闪点
闪点由成分中最低者决定
化学稳定性
副反应温度
隔膜
热收缩率
一般湿法隔膜热收缩率最低要求:纵向≤5%,横向≤3%
高温条件下,隔膜受热不均匀引发收缩变形造成电池内部短路。
①商用湿法PE隔膜两面涂覆Al2O3层可以有效地提高PE膜的吸液率和耐高温性能,在恒高温1h,涂覆膜收缩率最小。
热闭合温度
熔点
取决于材料的固有熔点
熔融破裂温度
粘接剂
热分解温度
LixC6和粘结剂的反应发生在250℃以上,放热量与粘结的种类及含量有关。放热量随着粘结剂含量的增加而增加。不同粘结剂的放热量不同。
熔融热
温度升高达到粘结剂的热分解温度,粘结剂分解放热。
自放电
正极
杂质
金属杂质
LiNiO2 K、Fe、Na、Ca、Cu、Cr≤0.03% wt
LiCoO2 Ni、Mg、Mn、Al、Fe、Na、Ca、Cu≤0.03% wt
LiMn2O4 K、Fe、Na、Ca、Cu≤0.03% wt
LiFePO4 Cr、Pb、Hg≤0.01% wt;Ge≤0.0005% wt
草酸亚铁路线合成的LFP,磁性杂质含量最高(1.63%),制成的电池自放电越大,是磁性杂质含量低(0.04%)的LFP电池的5.6倍
LiNixCoyMn1-x-yO2 Mg、Fe、Na、Ca、Zn、Cu、Si≤0.03% wt; SO42-≤0.5% wt;Cl-≤0.05% wt
金属杂质发生化学和电化学腐蚀反应,电池化成阶段电压达到金属元素的氧化还原电位后,金属就会先在正极氧化再到负极还原,当负极处的金属单质累积到一定程度,其沉积金属坚硬的棱角就会刺穿隔膜,在隔膜上形成黑点,导致正负极的微短路,不断消耗电量,导致电压降低。影响排序如:Cu>Zn>Fe>Fe2O3。(杂质含量高,自放电大;电池电压越高,自放电越大)
水分
水分造成电解液分解,释放出大量的电子,电子再嵌入到正极氧化结构中,从而引起正极电位下降,造成低压。
微粉
微粉颗粒大多为不完整的颗粒,颗粒表面缺陷多,增大了材料的比表面积,会加剧正极材料与电解液副反应的发生,也会增加微短路的几率。
比表面积
比表面积大 ,颗粒表面能升高,易团聚并加剧正极材料与电解液副反应的发生,也会增加微短路的几率。
负极
杂质
负极金属杂质会游离出来,在隔膜处沉积而造成隔膜导通,形成物理微短路,其中Cu、Zn对自放电影响较大。
金属杂质
磁性物质Fe+Cr+Ni+Zn<0.1ppm(0.00001%)
微粉
微粉颗粒大多为不完整的颗粒,颗粒表面缺陷多,增大了材料的比表面积,会加剧正极材料与电解液副反应的发生,也会增加微短路的几率。
比表面积
比表面积大 ,颗粒表面能升高,易团聚并加剧负极材料与电解液副反应的发生,也会增加微短路的几率。
天然石墨 2.0~2.2m2/g
人造石墨MCMB 1.6~2.0m2/g
软碳1.6~3.0m2/g
颗粒形貌
①负极材料表面不光滑,形成的SEI膜不稳定,导致存储过程中,SEI膜不断被破坏,负极继续与电解液发生副反应,形成新的沉淀修复SEI膜,如此循环,持续消耗电池中的活性锂,引起不可逆的自放电损失。 ②在电池存储过程中,负极中的电子会慢慢穿过SEI层与电解液发生副反应,使 SEI层不断变厚,造成负极材料中的活性物质减少。 ③低温条件下,随着SEI层不断变厚,负极表面会慢慢形成锂沉淀枝晶,这些枝晶继续发展会加重活性锂的流失,甚至会刺破隔膜造成内部短路,引发更严重的危害。
隔膜
表面缺陷
毛刺
针孔
隔膜表面存在针孔等缺陷,导致正负极接触,电子的转移路径是电解液,从负极通过隔膜到达正极,与正极材料发生还原反应,导致物理自放电。
孔径
隔膜孔径过大,导致正负极接触,泄漏电子造成的电池内部微短路,导致锂离子电池自放电。
机械强度
当隔膜机械强度不足,由于某种原因被破坏后,正负极接触,电子的转移路径是电解液,从负极通过隔膜到达正极,与正极材料发生还原反应,导致物理自放电。
PP 穿刺强度>0.204N/μm 拉伸强度-纵向 ≥100MPa 拉伸强度-横向 ≥25MPa
PE 穿刺强度>0.204N/μm 拉伸强度-纵向≥100MPa 拉伸强度-横向≥60MPa
电解液
杂质
水分
①有H2O存在时,其会与LiPF6反应,生产HF等腐蚀性气体;②在负极表面生成 LiOH沉淀,阻碍 Li+嵌入石墨,导致容量损失,同时与溶剂等反应产生CO2等气体引起电池膨胀。≤0.002% wt
HF
HF会与电池中众多物质如SEI反应,破坏SEI膜;生成CO2和H2O等;CO2引起电池膨胀,重新生成H2O又参与LiPF6、溶剂等反应,形成恶性链式反应。≤0.005% wt
金属杂质
金属杂质发生化学和电化学腐蚀反应,电池化成阶段电压达到金属元素的氧化还原电位后,金属就会先在正极氧化再到负极还原,当负极处的金属单质累积到一定程度,其沉积金属坚硬的棱角就会刺穿隔膜,在隔膜上形成黑点,导致正负极的微短路,不断消耗电量,导致电压降低。≤0.0001% wt
化学稳定性
溶剂不耐氧化,在存储过程中与正负极发生缓慢的化学反应,消耗容量而使得电压下降。
添加剂副反应
电解液中的氧气的存在会消耗活性锂并生成锂的氧化物,造成不可逆容量损失。
导电剂
金属杂质
金属杂质会游离出来,在隔膜处沉积而造成隔膜导通,形成物理微短路,其中Cu、Zn对自放电影响较大。
集流体
表面缺陷
油分
会导致涂布过程中材料与集流体接触不充分
表面损伤
腐蚀反应形成的铜离子能够在负极上重新沉积为金属铜,形成枝晶造成隔膜微短路,若枝晶继续发展将穿透隔膜,最终导致电池完全失效.
极片分切时,由于分切刀片不够锋利,会在极片边缘产生金属毛刺,这些毛刺会刺穿隔膜引发物理短路,导致自放电大的现象。
粘结剂
金属杂质
金属杂质会游离出来,在隔膜处沉积而造成隔膜导通,形成物理微短路,其中Cu、Zn对自放电影响较大。
隐性因素
SEI膜稳定性
一方面过渡金属离子溶解在电解液中并传导至电池负极,参与负极/电解液界面的SEI层的形成,这会增加SEI层的电导率,加快自放电的速度;另一方面过渡金属离子的溶解也导致正极活性材料减少,造成不可逆容量损失。
①负极中的电子会慢慢穿过SEI膜与电解液发生副反应,SEI膜不断变厚,负极表面慢慢形成锂沉淀枝晶,枝晶继续发展会加重活性锂的流失,甚至会刺破隔膜造成内部短路,引起不可逆自放电;② 锂离子、碳以及电解液生成的 LiF、Li2CO3、以及其他聚合物等造成了电池的不可逆容量损失。SEI膜破坏的后果:①溶剂进入石墨层中与LixC6反应,引起不可逆容量损失;②破坏的SEI修复则要消耗Li+和溶剂等,进一步造成不可逆容量损失。
过度金属溶解
①电解液在界面被氧化分解,并在正极表面形成富含锂的SEI层。LCO或NCM的SEI层易出现裂痕,使正极与电解液再次接触,不断进行界面副反应,造成不可逆自放电增大;②采用LiPF6作为电解质的电池,经过短期高温冲击后,SEI层中形成的LiP3可不断地向正极结构中嵌入Li+,诱发显著的自放电现象;
电化学体系窗口
LiPF6 LiPF6热稳定性差,60~80℃左右就有少量分解成为LiF
LiAsF6 对碳负极电化学性能最好,但其毒性较大
LiClO4 氧化性太强,安全性差