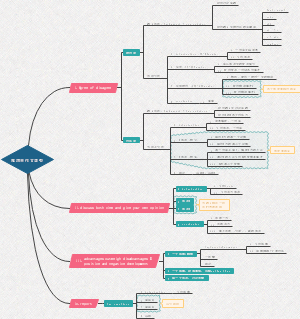
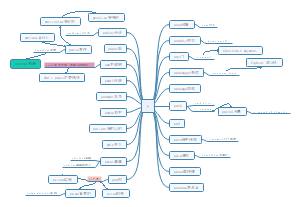
Growth factor receptors will generally activate Ras protein
Different phosphorylated residues can recruit different protein adaptors and may trigure 3 different signaling pathways, the most important pathway we talk among those 3 is Ras
3 pathways are linked under Ras:
1. MAPK pathway induces cell growth and shape change
2. PI3K pathway induces the evading of appoptosis
Here is an alternative treatment of the membrane bounded lipid PIP2
phospholipase C will cleaves phosphodiester bond of PIP2 to yield IP3(Free part) and DAG(Membrane bounded part)
PI3K first phosphorylates PIP2 into PIP3, and PIP3 will recruits Akt/PKB, PDK1 and PDK2 will phosphorylate and activate the PIP3 bounded Akt/PKB, the activated Akt/PKB will transduce its signal to the downstream
the docking domain of AKt/PKB on PIP3 is called PH domain.
A phosphate called PTEN here provides a negative feedback mechanism, it dephosphorylates PIP3 back into PIP2 and inhibits the downstream signals.
PTEN is a tumour suppressor protein.
The loss of function mutations on PTEN are involved in many kinds of cancer.
In human tumours, the transcription of PTEN often get inhibited through the methylation on its promotor region.
3. Ral-GEF pathway induces the change of cell mobilities
Incidences of oncogenesis
1. Over expression of Growth factor receptors induce hyper responsiveness of growth factor.
Most tumours over express EGFR
1. mostly increase transcriptional level
2. some may be amplification of genes
3. some may decrease Protein degredation/decrease endocytosis, endocytosis regulates the life time of cell surface proteins.
4. viral encoded growth factor homologous
v-sis viral derived oncogene of siman sarcoma virus is the homologous of the B chain of platelet-derived groth factor protein(PDGF)
viro oncoproteins can be constitutively activated EGFR
v-ErbB encodes a EGF-R homologous but lack part of the ectodomain, which can release mitogenic signals constitutively
3. caused by gene fusion mutations (induce dimerization)
part of N terminus Muscle tropomyosin replaces part of the N terminus extracellular domain of Trk, the resulting oncoprotein is constantly dimerized and constitutively activated, and it locates only in cytosol.
3. Disruptions of negative-feedback mechanisms that attenuate proliferative signaling.
4. Over expressions of self-effective growth factors,which cause Autocrine and paracrine stimulation of cell growth.
signals that effect on its own.
This leads to self-sufficiency in growth factors, which finally reduces its dependence on the growth factors in its micro enviroment.
The symptom is similar with the over expression of growth factor receptors.
therapeutic targets
Monoclonal antibodies against growth factor receptors.
Kinase inhibitors inhibits key signaling kinases.
Cell adhersion receptors
Integrins
hetero dimeric structural organization
α plus β subunit。
Outside in signaling
When ectodomains bind to specific components in EM, intermediary proteins will link the cytoplasmic domain of beta subunit to the cytoskeleton(actin fibires). at the same time, the cytoplasmic domains of beta subunit can attract variaty of signaling molecules.
The signaling adaptors of integrins
FAK(focal adhesion kinase)
It can activates most pathways activated by growth factor receptors.
inside out signaling
cytoplasmic signals can control the binding affinities of integrins for their ECM ligands.
this may lead to the breaking existing contacts and forging new ones in their place.
roles in cell motility
inside out signalings induce integrin to forge new linkages with the ECM.
When cell needs to loosen its tehers at the front, signalings will cause release contact with the substratum.
E-cadherin
Intermediate proteins(p120, β-catenin, α-catenin) link cadherin's intracellular domain with cytoskeleton(actin filament).
β-catenin
The accumulation of β-catenin is very common in cancer.
as a transcription factor, it can direct transcriptional express certain proteins.
axin -> GSK-3β(glycogen synthase kinase-3β) +p-|(targets it for degredation) β-catenin
Apc (Adenomatous polyposis coli) here is part of the protein complex: {axin][GSK-3β][APC][β-catenin][Wtx}
it is critical for the successfuly capture of β-catenin, that is then phosphorylated and targeted to degredation by β-catenin.
Onco-proteins in and around β-catenin
Constitutively activated β-catenin.
APC mtants(fail to bind and down regulates β-catenin levels).
Cell cycle checking motif
pRb
pRb A molecular checkpoint for the cell cycle restriction point
pRb -| E2F -o-> Proteins to path restriction points
It is phosphorylated by CDKs
Cyclin D-CDK4/6 +p pRb -|-> E2F -o-> DNA polymerase and other proteins for DNA synthesis
interpretation: pRb is the checkpoint to certain stage of cell cycle, because it normally inhibits cell cycle, and it needs to be induced by CDK2-CyclinE.
It also contains a design of positive feed back loop
E2F -o-> Clyclin E, CDK-2, E2F
CyclinE-CDK-2 +p pRb -|-> E2F -o-> ~~~~
So we call it a positive feed back loop
The phosphorylation of pRb becomes CyclinD independent.
Pictures
in cancer, pRb is often inactivated by lose of function mutations.
TGFβ
TGFβ is the receptor for antigrowth signals, TGFβ activation inhibits the cell cycle progression
Type I TGFβ signaling
TGFβ -> SMADs --o-> p15, p21
Here
p21 is also transcirptional activated by p53
p15 has a full name of p15INK4b
p15 -| CyclinD-CDK4-6 --> path of restriction point
p21 -| CyclinE-CDK2 --> path of restriction point
TGFβ -> SMADs -o-| c-Myc -| Miz -o-> p15ink4b,p21zip -| cyclin-CDKs -> path of restriction points.
p.s.
an error in the picture, p15ink 4b and p21zip has the same roles with p15 and p21, i.e. they inhibit cyclines to arrest cell cycles
TGFbeta
Type II TGFβ signaling
in cancer, TGFβ is often inactibated by lose of function mutations.
SMADs(the protein adaptors at down stream of TGFβ) is often inactivated by lose of function mutations.
smad2- in colon cancer
smad4- in pancrease cancer
c-Myc
cMyc is another cell cycle check point protein, it is a transcroptional factor that can either inhibits or activates cell cycle progression depends on its interacting partners
c-Myc can dimerizes with
c-Myc(form homodimer)
Max
Myc-Max is a transcriptional activator
Myc-Max -o-> Cyclin D2, E2F1, 2,3 , CDK4
results: cell will path G1 checkpoint
Miz
Myc-Miz can be thought as a transcriptional repressor
Miz is a transcriptional activator, and Myc binds and inhibits Miz.