导图社区 六上数学(部分)
上海六上数学(部分)的思维导图,主要内容有整数、整除、因数、能被2、5整除的数、质数、合数、分解素因数等。
社区模板帮助中心,点此进入>>
《稻草人》读书笔记
《童年》读书笔记
《昆虫记》思维导图
《安徒生童话》思维导图
《鲁滨逊漂流记》读书笔记
《春晓》思维导图
亡羊补牢
一张思维导图帮您读懂唐诗《咏鹅》!
外婆与姥姥的区别
父母学吧四大模块课程
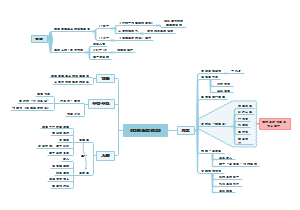
六上数学(部分
整数
零和正整数统称为自然数。正整数、零、负整数,统称为整数
整除
整数a除以整数b,如果除得的商视整数而余数为零,我们就说a能被b整除;或者说b能整除a。
因数
整数a能被整数b整除,a就叫做b的倍数b就叫做a的因数。
能被2、5整除的数
能被2整除的整数叫做偶数,不能被2整除的整数叫做奇数。个位上是0或者5整数都能被5整除。
质数、合数
一个正整数,如果只有1和它本身两个因数,这样的数叫做素数,也叫做质数,如果除了1和它本身以外还有别的因数,这样的数叫做合数。
分解素因数
每个合数都可以写成几个素数相乘的形式,其中每个素数都是这个合数的因数,叫做这个合数的素因数,把一个合数用素因数相乘的形式表示出来,叫做分解素因数。
最大公因数
几个整数公有的因数叫做这几个整数的公因数,其中最大的一个叫做这几个整数的最大公因数。
最小公倍数
几个整数公有的倍数叫做这几个整数的公倍数,其中最小的一个叫做这个几个整数的最小公倍数。
互素
两个正整数中,如果某个数是另一个数的因数,那么这个数就是这两个数的最大公因数,如果这两个数互素,那么它们的最大公因数就是1。
分数
两个正整数p、q相除,可以用分数p/q表示,p÷q=p/q,其中p为分子,q为分母。
分数的基本性质
分数的分子和分母同时乘以或者除以相同的数(0除外),分数的大小不变,即a/b=am/bm=a÷n/b÷n (b≠0,m≠0,n≠0)
最简分数
分子和分母互素的分数,叫做最简分数。
约分
把一个分数的分子与分母的公因数约去的过程,称为约分。
通分
将异分母和分数分别化成与原分数大小相等的同分母的分数,这个过程叫做通分。
分母的计算
异分母分数相加减,先通分,然后按照同分母分数相加减的法则进行计算。分子比分母小的分数叫做真分数,分子大于或者等于分母的分数叫做假分数。一个正整数与一个真分数相加所成的数叫做带分数。