导图社区 治疗用生物制品病毒污染风险控制要点
治疗用生物制品病毒污染风险控制要点
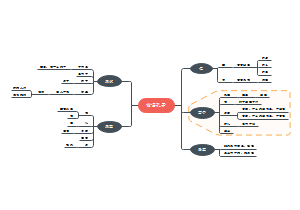
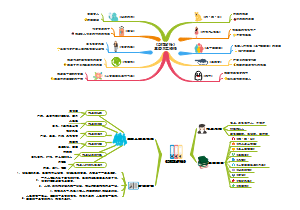
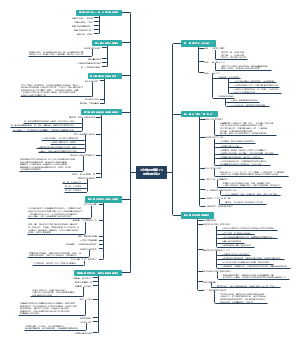
治疗用生物制品病毒污染风险控制要点总结,包括生物制品病毒清除/灭活策略,生产检定用细胞的质控策略等等。
提示: 本内容由社区用户上传并分享。平台不对内容的真实性、合法性、知识产权归属及是否侵害第三方权利进行事前审核或保证。本内容可能包含受版权保护的图片、字体或其他第三方素材,使用前请自行确认授权范围。
- 生物制品
- 细胞检定
- 病毒灭活
- 病毒清除
- 外源病毒因子
他的近期作品
查看更多>>
- CAPA(纠正和预防措施)实施的八大流程
这是一篇关于CAPA(纠正和预防措施)实施的八大流程的思维导图,主要内容包括:引言,CAPA实施的八大流程,CAPA实施中的常用质量工具。
- GLP一般毒理研究试验方案的组成
这是一篇关于GLP一般毒理研究试验方案的组成的思维导图,主要内容包括:遵守良好实验室规范(GLP)声明,计算机化系统,基本信息与研究目标,受试物相关,试验系统和动物相关,动物选择与处理,试验分组与处理,特殊组别与评估,试验后评估,其他补充部分。
- 治疗用生物制品病毒污染风险控制要点
治疗用生物制品病毒污染风险控制要点总结,包括生物制品病毒清除/灭活策略,生产检定用细胞的质控策略等等。
治疗用生物制品病毒污染风险控制要点
社区模板帮助中心,点此进入>>
他的近期作品
查看更多>>
- CAPA(纠正和预防措施)实施的八大流程
这是一篇关于CAPA(纠正和预防措施)实施的八大流程的思维导图,主要内容包括:引言,CAPA实施的八大流程,CAPA实施中的常用质量工具。
- GLP一般毒理研究试验方案的组成
这是一篇关于GLP一般毒理研究试验方案的组成的思维导图,主要内容包括:遵守良好实验室规范(GLP)声明,计算机化系统,基本信息与研究目标,受试物相关,试验系统和动物相关,动物选择与处理,试验分组与处理,特殊组别与评估,试验后评估,其他补充部分。
- 治疗用生物制品病毒污染风险控制要点
治疗用生物制品病毒污染风险控制要点总结,包括生物制品病毒清除/灭活策略,生产检定用细胞的质控策略等等。
- 相似推荐
- 大纲