导图社区 核酸实时定量检测技术
核酸实时定量检测技术
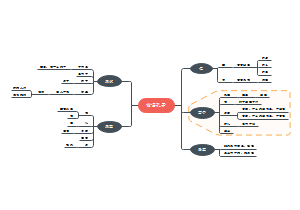
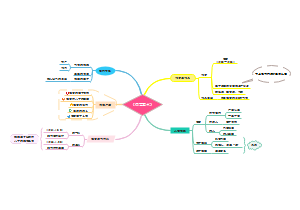
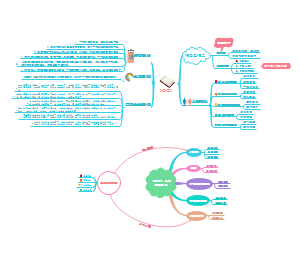
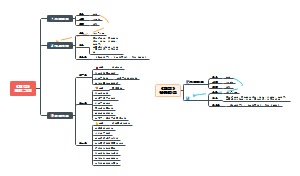
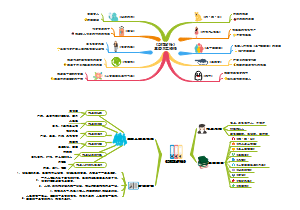
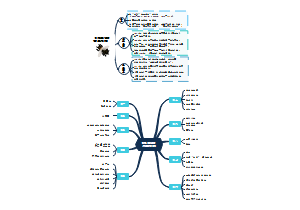
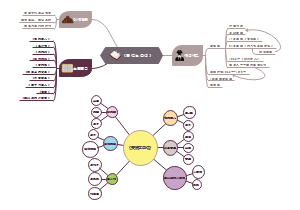
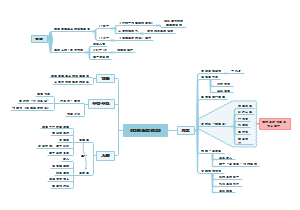
临床分子生物学检验技术第六章,内容有实时荧光定量PCR技术(Q-PCR)、Q-PCR的基本原理、Q-PCR引物和探针的设计、Q-PCR反应体系和条件的优化、Q-PCR测定的数据分析。
编辑于2023-09-23 11:06:01 四川省- 医学检验技术
- 相似推荐
- 大纲
导图社区 核酸实时定量检测技术
临床分子生物学检验技术第六章,内容有实时荧光定量PCR技术(Q-PCR)、Q-PCR的基本原理、Q-PCR引物和探针的设计、Q-PCR反应体系和条件的优化、Q-PCR测定的数据分析。
编辑于2023-09-23 11:06:01 四川省