导图社区 All mechanis-ms involved in fheling test
- 25
- 0
- 0
- 举报
All mechanis-ms involved in fheling test
All mechanis-ms involved in fheling test思维导图,包括:hydrolysis of disaccharide/glycosides、monosacaride's ring open、interconvertion between chain ketose and aldose等内容。
提示: 本内容由社区用户上传并分享。平台不对内容的真实性、合法性、知识产权归属及是否侵害第三方权利进行事前审核或保证。本内容可能包含受版权保护的图片、字体或其他第三方素材,使用前请自行确认授权范围。
- Oxidation
- hydrolysis
- mechanis…
- Cu2
- disacchari…
- 相似推荐
- 大纲
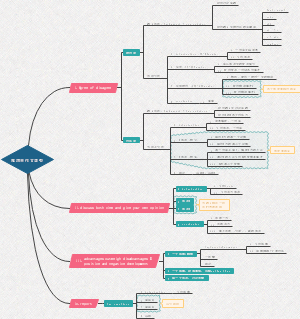
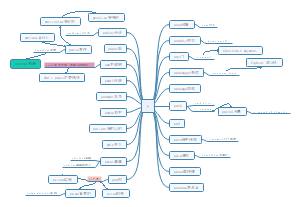

All mechanisms involved in fheling test
hydrolysis of disaccharide/glycosides
reverse of acetyl formation
catalyzed by acid
equilibrium moved backward by adding excess water
highly reversible
basically all steps
their structures don't show much difference in energy
very slow in water/basic solution
ex. sucrose water solution remain stable for years
monosacaride's ring open
reverse of hemiacetyl formation
catalyzed by acid
both rate and equilibrium state are effected by energy of ring structure
because the free energy change and activation energy all different.
their transition state's energy are similar
β-D glucose is most stable state
no axil steric string
minimized equitorial steric sting
no tortion strain
no angle starin
effect
established most in equilirium
99.
5 membered ring(furanose) is not as stable
highly reversible
ring doesn't seem to be stabalized that much
but also significant more stable
>99% in equilibrium
basically all step
slow, should be rate limiting
explain our observation
more quantification using standard Cu2O curve should be done to measure the rate accurately
interconvertion between chain ketose and aldose
keto-enol tautomerism
catalyzed by acid
followed by E1 like deprotonation
not accelerated by stonger nucleophile added
Through cation intermediate
intermediat is majority in equilibrium if enough acid is applied
Keq not change, equilibrium move to middle because excess amount of both "reactant"
Subtopic
catalyzed by base
Through Enolate ion intermediate
intermediate is majority in equilibrium if enough base is added
Keq not change, equilibrium move to middle because excess amount of both "reactant"
Subtopic
2 continous tautomerisms happened to make one event of ketone/aldehyde interconversion
highly reversible
equilibrium normally exist in water solution, but generally very little enol concentration in equilibrium due to its high energy/less stable structure.
fast
our observation in base
not in neutral
Oxidation of aldose by Cu2+
general aldehyde oxidation
1. hydration of aldehyde group into a 1,1-diol
nucleophilic addition
catalyzed by both acid and base
2. oxidant substitutes one hydroxyl hydrogen
3. elimination of the metal and another hydrogen on C1.
E2 close related
therefore accelerated by better nucleophiles applied
4. generate reduced metal and carboxylic acid
basically irreversible
oxidation has high free energy change
e- is stabalized by higher electronegativity atoms
fast
by observation
we have catalyst
It is proved that through enolate ion intermediate
Alkaline cleavage of monosaccharides
reverse reaction of aldol condensation reactions
1. base/OH- is an essential catalyst
excess base make equilibrium move to tetrahedral alkoxide ion intermediate + enolate ion with the aldehyde/ketone
2. reverse reaction is enhaced by divalent calcium, by it complexation with the aldol product
2. inhibited if aldehyde is oxidized into carboxylic acid
acid is hard to generate enolate ion
acid enolate ion is not stable due to repelling of 2 negative charged oxygen(high energy dianion intermediate)
can be coordinated by a divalent metal cation as a lewis acid, make is more stable
acid's cation intermediate of acid catalzed keto-eno tautomerism is higher in energy
the carbocation is very high in energy because the inductive effect of 2 oxygen directly connected to it
reversible
low free energy change
effected by condensation reactants' structures
favor condensation product
aldehyde with no α-substitution: RCH2CHO
favor cleavage product
disubstituted aldehyde :R2CHCHO
Most ketones
steric factors are most important
increased substitution near the reaction site increases steric congestion in the adol product
make highenergy aldol product
so the equilibria favor cleavage
fast
equilibrium normally fast reached in NaOH with protic solvent
but slow when PH<7
Important concepts to discuss kinetics
catalyst
only lower activation energy!
through another route! so must require intermediate !
So might effect the yield of product by producing to much intermediate
ex. catalytic amount of NaOH will favor aldo condensation.
0.05 equivalent
thermaldynamic favor
warm environment
1 full equivalent of NaOH, will favor the α-substitution with other electrophiles
generate all enolate ion
kinetic favor
cold environment
only reduce time to reach equilirium!/never change equilibria
but the proportion of reactant/product never change
determined by keq
do not change on Keq/equilibria
means the proportion of the most left(reactant) and most right(product) never change
But might effect the yield of product by producing to much intermediate
enzymes
excess enzymes never produce any intermediate
under standard modle,The step intermediate converted to product: ES---->E+P is inreversible
because its very high selectivity through molecular recognation
but it's reversible of most inorganic catalyst
ex.base can generate enolate ion
and catalytic amount of base use enolate ion as intermediate to increase rate of keto-eno tautomerism
enzyme catalyst will dramatically change equilibria
It only dramaticaly increase the rate forward but not the rate backward
you can't back from the same "hill" that you gone
never in other catalyst
non-equivilant reverse
never specific acid/base catalyzes but general acid/base catalyzes
specific involve multisteps
general usually only one step
Specific/general acid/base catalyst
specific
have intermediate
means isolatable/ finite life time protonated/ deprotonated form
only under strong PH
general
no intermediate
under moderate PH like most biological system's reaction
Move of equilibrium
by changing concentration of any molecules involved in the reaction
no change on Keq
That's why we have keq, just inorder to reason the concentration's proportion/ because proportion of those should finally be a constant in one movement state(temperature)
by defination no standard delta G and temperature changes, no Keq changes
standard delta G changes only when you change the structure of molecules/ or you change the reaction you are talking about
by changing temperature
change on Keq
by changing pressure
change on Keq
The direction of movement always against the change
methamatical tools
Equilibria(K)
determined by standard free energy change and temperature △GΘ=-RTlnKeq
the energy state of reactant/product
if you don't change the reaction/process you are talking about, then the △GΘ will not change at all
exponentially
means when temperature not changed, equilibria is sure as long as we know the accurate energy differences
The funcion allow you to modulate any temperature
When T→∞
Keq→1
Weaken the effect of△GΘ
so increased temperature makes exergonic and endergonic do not make too many differences in final proportion
Another important formular: ΔG=ΔGΘ+RTlnQ
When ΔG=0, Q=Keq
Define in any instances, the movement forces of a reaction
It derives the upper formula
It's derived from △G=△H-T△S
RlnQ is part of ΔS
K is the Q when equilibria
Q defined by[product]^n/[reactant]^n
there for can anticipate the proportion of reactant/product in final equilibrium
weakness: equilibria only tell you the proportion but not the actual concentration, you can't know intermediate's proportion
further question: how to reason intermediate here?
you can calculate any Keq if you know △G between any 2 substances, they can be considered as a product/ reactant, eventhough they are actually intermediate, and their concentration's proportion is also fixed during equilibria
pKa is the individual reverse step's Keq
Keq usually is talking about start and end
that's how catalyst can effect the final proportion of yield
doesnot change equilibria/keq/proportion of two sides, but increase the intermediate amount
therefore if you don't diliberately want that intermediate, catalyst's amongt shouldn't be too much
ex. catalyst amont of OH- in aldose reaction
we don't want enolate ion but let it as an intermediate to catalyst
vs 1 full equivalent of OH- in enolate ion production
then happen α-substitution
we want enolate ion
ΔG between seady states, not the lowest energy state
so intermediate could be also significant to effect final yields
heve other names lile K,Ka,Kb
They are Q in equilibria
Q is part of entropy
reversible/irreversible is equilibria question
Keq>>1
irreversible
Keq around 1
reversible
imagine balls fall into a very very deep hole and it cannot jump out back again
Rate(k)
determined by k
determined by activation energy
determined by energy different between steady state and transition state
more accurately difference between start state(steady state) in reaction coordinate and the highest energy state(rate limiting step's transition state)
transition state
peaks on reaction coordinate
1/2 probabilities to roll forward and back ward
if there are multiple steps. catalyst must act on the rate limiting step to increase the rate
weakness of rate limiting step idea
Only applied to multi step reactions with significant transition state energy differences/ on significant higher than others
my opinion: the criteria for significant
e.x. in a 2 steps reaction, if the second step is rate limiting, then the △Gact 2- △G act1 must be always bigger than △GΦ for step 1
their can be multi rate limiting steps in a single reaction site, because they are almost same in energy.
Hammond postulate
The transition state of an exergonic reaction step structurally resembles the reactant, whereas the transition state of an endergonic reaction step structurally resembles the product.
exponentially
Temperature
exponentially
and concentration of reactant(s)
first order
V=k[S]
unit of k: S^-1
ex. SN1/E1
2 steps, the first step is rate limiting, overall first order kinetics, only depends on [alkyl halide]
second order
V=k[S1][S2]
unit of k: S^-1*M^-1
ex. SN2/E2
1 step, depends on both[alkyl halide] and [nucleophile]
△G/free energy it self
△G=△H-T△S
H: enthalphy/toal energy~ all the kinetic energy+potential energy in the system。
in C nucleus
Calculated by E=mc^2
2*10^9 kcal/mol
in 2 core electrons of C(1s)
2*10^4 kcal/mol
measured by putting them to C core
C 4 valence electrons 2s+2p
3*10^3 kcal/mol
correlation energy: mistakes of best calculation we have(MO)
1*10^2 kcal/mol
in covalent bonds
1*10^2 kcal/mol; C-C bonds
in noncovalent contancts
1-20 kcal/mol
total conformation energy
in bond streching
angle strain
torsional strain
van der waals( steric strain)
When nonbonded atoms that approach each other too closely
intermolecular interactions( van der waals force)
permanent& regular dipoles: dipole-dipole forces
attractive geometry
Usually predominants cause lower in energy
Repulsive geometry
between polar molecules
Transient dipoles: dispersion forces
between all neighboring molecules
because the electron distribution with in molecules is constantly changing
transient dipoles are formed
why substances are liquid and solid, the comulitive effect is strong
increase when molecules are bigger, and more surface can aligned each other
ex. fatty acid 's melting point
special dipoles: hydrogen bonds
hydrophobic
hydrophilic
correlation interactions between He*He
2*10^-6 kcal/mol
not determined
So enthalpy means all the real energies, which changes must reflect in heat (mostly) or other forms (transformation)
first law of thermal dynamics define H, and its behavior
S: entropy---the freedom of motion
The S is the amount of energy necessary for particles used to maintain the absolute temperature
So more freedom of motion, more proportion of the energy will used to maintain the temperature (speed of motion)
also higher speed of motion/average kinetic energy of motion, the higher energy it cost for the total motion
So why T*ΔH
Temperature is really a statistical idea
"Average" kinetic energy
entropy is likely to be more solid
concentration
concentration gradience in diffusion
Q
partern of reaction
Q
Intramolecular fixation/ free spaces
ex. the intramolecular catalyst case
totally is the motional freedom of the system you talk
it is not real a form of energy, but change of it can be part of free energy barrier..
energy can't used for purpose/ energy barrier to a reaction(activation energy)
More direct kinetic view: because the probability of productive collision is lowered by motion's freedom/disorder
can be overcomed by enzymatic catalysts
lower transition state's energy by essentially tricky process/lower their order
imagine, transition state's energy G is really high if a transition state required is very order, so activation energy is very high
Can be inter/intra molecular
if more, molecules must spent more energy to fight against it
Because molecules need maintain the temperature!
So higher the energy, higher the energy it required
absolutely independence of Temperature
determained by molecules design/process it self
Dr.T is absolutely wrong
part of enthalpy changes depends on the concentration and their proportion also equation(Q)
Q
Keq is the Q when equilibrium
means under cirtain temperature, the Q build up when equilibrium is always the same.
The remaind entropy seems constant: ΔSΘ
measured under standard condation
classicle modles
ΔS>0
diffussion
ice melting
ΔS<0
water freezing
In all nature processes, entropy of the universe increase
second law of thermaldynamics
T: absolute temperature
absolute temperature is proportional to the average kinetic energy of the random microscopic motions of their constituent microscopic particles such as electrons, atoms, and molecules.
only when P/V is not changing
biological condation never changes
ΔG=ΔGΘ+RTlnQ
Activation energy/ energy barrier constists
1. entropy
probability of productive collision(correct orientation) is lowered by motion's freedom/disorder
majority
over comed by presice of enzyme
2. enthalpy
like solvation shell of water require to be overcomed in most biological process
this factor also contributes to entropy..
Stuctural/enviromental stabilization of transition state
steric hinder
Whatever
under nonstandard condation is hard to accurately quantification, but it still works to tell you the general direction and scale of changes
summary
temperature effects on both k and keq
But Keq and freeenergy change doesn't direct determine rate.. you should look at transition state's energy to determine the rate
always think about sucrose and diamond
or just 2 things
rate/kinetics: k
look at activation energy
rate limiting step's transition state's energy
catalyst can act on it by providing alternative route
but it's not even indirectly correlate with△G
So doesn't effect equilibrium
further question:look at how to test it
determine how fast it happens/ reach the equilibrium
equilibria/thermodynamics: K
look at standard free energy difference
depends on reactant and product's energy state
determine whether the reaction can happen or not/spontaneous or not
if it can:reversible? or irreversible
when 2 reactions compete with eachother
thermodynamic favor
refer to the reaction has lower k, but more stabalized product(more negative △G) than the other
usually by heating to help this reaction
make it equilibrium establish faster
you can also selectively catalyst it to make it both favor
kinetic favor
refer to the reaction has higher k, but less stabalized product(less negative △G) than the other
usually by avoid heating/low temperature to help it to win the competation
because finally it won't compete with the thermal dynamicaly favored one if you don't isolate its product
both favor
so the other both not favor
genrally working
you can always isolate special product of one reaction to increase one reaction's yield
you can always inhibit one reaction by not providing/isolate its reactant
Remember, when you think about free energy
It's defined by a process/ a reaction
not particular molecule! but the molecule in this situation
You can't define G of each molecules
it's correlated with molecules' s total energy/like bonding energies inherited its structure, but you can't know ΔG, unless you experimentally precisely measure the probability of collition/ freedom of motion changes/ the enthalpy thing
H is possible to find
But how to define and measure S?
and how about each molecules account for?many reaction has more than 1 reactant/product
or only in one reaction(so you can use reaction coordinate), another different process the molecule's G will definately change
This is the feature of chaos/dynamic system
but you can add processes's ΔG under a common standard condation
Reaction coordinates are great tools
but always remember one "valey" may represents 2 molecules in a second order process/step
Classic condations to reason
melting point
the point of ΔH = TΔS
for crystal the ΔS is very big and constant
for noncrystal the ΔS is changing every step, hard to define, so no melting point
so under a define T
diffusion
it selt drive completely by negative TΔS
ΔH=0
solvation
more hydrogen bonds are formed, ΔH<0.。less potential energy by new noncovalent interaction forms
maximize hydrogen bonding
solubility
more complex