导图社区 七上地理
七上地理
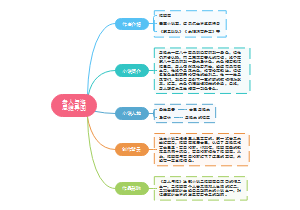
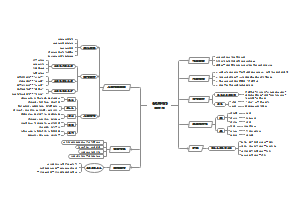
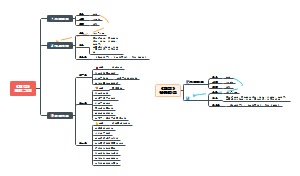
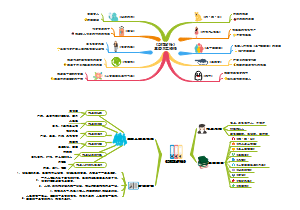
这是一个关于七上地理的思维导图,中国的地势与地形:西高东低的地势,对农作物、森林、牧草的生长极为有利,西高东低的地势,方便了沿海与内陆的经济联系,蕴含了丰富的水能资源。
编辑于2022-02-13 10:35:44- 七上地理
- 相似推荐
- 大纲
导图社区 七上地理
这是一个关于七上地理的思维导图,中国的地势与地形:西高东低的地势,对农作物、森林、牧草的生长极为有利,西高东低的地势,方便了沿海与内陆的经济联系,蕴含了丰富的水能资源。
编辑于2022-02-13 10:35:44