导图社区 SNP实战生物信息学变异检测callsnp
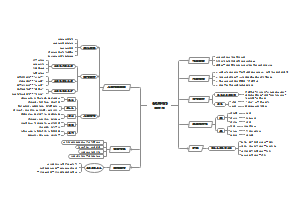
变异信息提取(callSNPpipeline)1(oldversion) 群体遗传大部分的分析大部分基于VCF文件,所以得到一个高质量的VCF文件很有必要。
社区模板帮助中心,点此进入>>
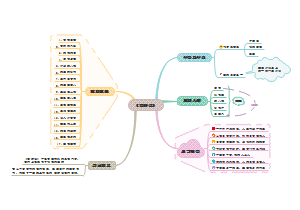
《老人与海》思维导图
《钢铁是怎样炼成的》章节概要图
《傅雷家书》思维导图
《阿房宫赋》思维导图
《西游记》思维导图
《水浒传》思维导图
《茶馆》思维导图
《朝花夕拾》篇目思维导图
英语词性
生物必修一
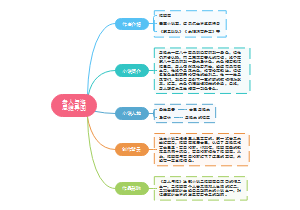
call snp
提取VCF中有用信息并以200或500为窗口进行统计
VCF信息提取,提取1、2和个体对应的列(位于10以后),生成SNP文件
以自定窗口数目进行统计,使用的是heter_num_ratio_cal-progeny.py,生成ratio.txt文件
若有多个个体
将个体ratio.txt文件合并,生成merge文件,可用excel展示
用R画成PDF
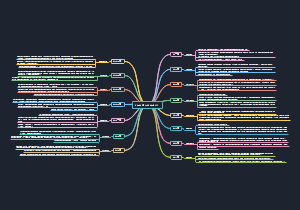
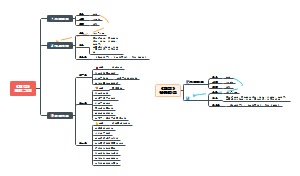
bcftools mpileup -Ou -f genome.fa S.sort.rmdup.bam | bcftools call -mv -Ou | bcftools filter -s LowQual -e ‘%QUAL<30 || DP <8 || MQ <35 || DP > 80’ > S.vcf
命令意义
mpileup
变异检测,生成初始的VCF文件,与call连用
call
进行SNP/INDEX检测
m是新版变异检测参数,与旧版c不得一起用
v(小写)是只输出有变异的位点,V(大写)是忽略SNP/INDEX位点
O(大写)规定输出文件的格式
u表示未压缩的BCF
b表示压缩的BCF
z表示压缩的VCF
v表示未压缩的VCF
Ou连用,可以减少VCF与BCF的转换,加速处理过程
filter
-s LowQual 将文件注释为 LowQual
-e
常与-s连用,满足-e后面的条件被排除,标记为PASS;不满足的被留下,传递到filter标记为LowQual
QUAL
Quality变异质量值,值越大变异可能越大
DP
Depth 测序覆盖深度
MQ
所有比对至该位点上的read的比对质量值的均方根(先平方、再平均、然后开方)一般好的位点MQ为60左右
||
用于分割命令,前一个命令为假,才会进行下一个命令
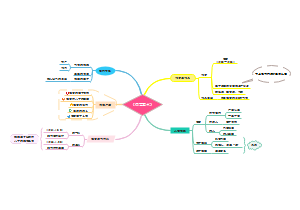
picard -Xmx4g MarkDuplicates I= S.sort.bam O=S.sort.rmdup.bam CREATE_INDEX=true REMOVE_DUPLICATES=true M=S.marked_dup_metrics.txt
生成的 .bam / .bai 保存一处
bwa mem -t 1 genome.fasta S1.fa.gz S2.fa.gz | samtools sort -@ 2 -m 1G -o S.sort.bam
bwa index genome.fasta
参考基因组Reference genome
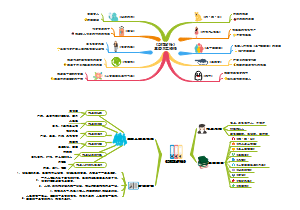
SNP实战
454-10的SNP位点开始
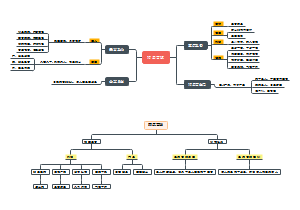
对 A056/C151/C454三个亲本(C151是A056的姊妹系)进行变异检测
以染色体为单位进行Call SNP ,使用 for in 遍历,一步到位
对检测生成的VCF进行提取
提取出符合我们要求的位点后,进一步进行过滤
生成的SNP文件,用于下一步的20个体的Call SNP
使用 -b插入包含了20个体的路径的文件,最后生成初始的raw,vcf文件
257645 个亲本SNP位点,20个list 中154554个Call SNP位点
200window
500window
对A056/C454两个亲本进行变异检测
4个条件
亲本SNP316095,20个list中182596个SNP
3个条件
亲本SNP281246,20个list中147742个SNP
500
200
2个条件
亲本SNP175731,20个list中126624个SNP