导图社区 NMPA:除菌过滤技术及应用指南-2018
NMPA:除菌过滤技术及应用指南-2018
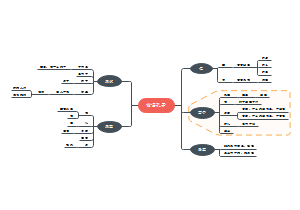
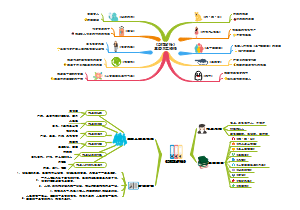
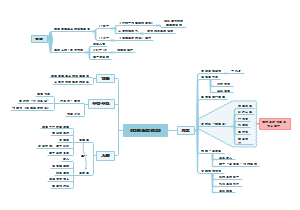
通过该思维导图可以快速了解该指南的要求。本指南旨在规范除菌过滤技术在无菌药品生产中的应用,主要目标是确保药品的安全性、有效性和质量稳定性。除菌过滤技术是通过物理截留的方法去除液体或气体中的微生物,以满足无菌药品相关的质量要求。保证药品的安全性、有效性和质量稳定性。
编辑于2024-06-15 13:56:25- 制药行业
- 验证知识
- ECA Good Practice Guide 确认与验证 3.0
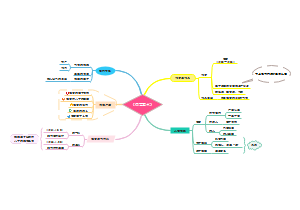
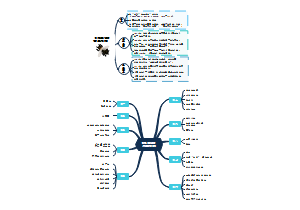
这是一篇关于ECA Good Practice GuideQualif的思维导图,主要内容包括:8 Support by Use of Electronic Documentation使用电子文档提供支持,7 Support by Categorisation 按分类提供支持,6 Supplier Activities供应商活动,5 Customer Activities客户活动。
- NMPA:除菌过滤技术及应用指南-2018
通过该思维导图可以快速了解该指南的要求。本指南旨在规范除菌过滤技术在无菌药品生产中的应用,主要目标是确保药品的安全性、有效性和质量稳定性。除菌过滤技术是通过物理截留的方法去除液体或气体中的微生物,以满足无菌药品相关的质量要求。保证药品的安全性、有效性和质量稳定性。
NMPA:除菌过滤技术及应用指南-2018
社区模板帮助中心,点此进入>>
- ECA Good Practice Guide 确认与验证 3.0
这是一篇关于ECA Good Practice GuideQualif的思维导图,主要内容包括:8 Support by Use of Electronic Documentation使用电子文档提供支持,7 Support by Categorisation 按分类提供支持,6 Supplier Activities供应商活动,5 Customer Activities客户活动。
- NMPA:除菌过滤技术及应用指南-2018
通过该思维导图可以快速了解该指南的要求。本指南旨在规范除菌过滤技术在无菌药品生产中的应用,主要目标是确保药品的安全性、有效性和质量稳定性。除菌过滤技术是通过物理截留的方法去除液体或气体中的微生物,以满足无菌药品相关的质量要求。保证药品的安全性、有效性和质量稳定性。
- 相似推荐
- 大纲