导图社区 ISO13485-2016医疗器械质量管理体系要求
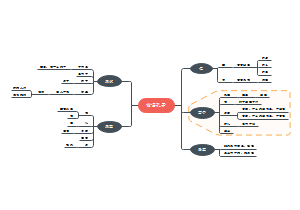
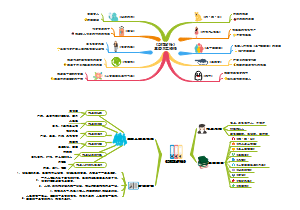
ISO13485-2016医疗器械质量管理体系要求
包括了组织应策划和控制产品的设计和开发。适当时,随着设计和开发的进展,应保持和更新设计和开发计划文件。
提示: 本内容由社区用户上传并分享。平台不对内容的真实性、合法性、知识产权归属及是否侵害第三方权利进行事前审核或保证。本内容可能包含受版权保护的图片、字体或其他第三方素材,使用前请自行确认授权范围。
- 医疗器械
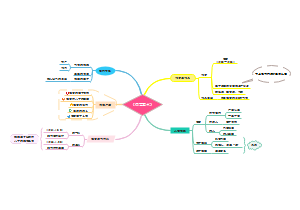
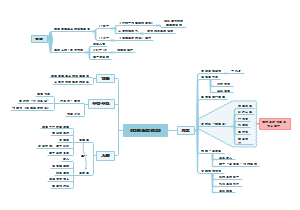
- 公立医院运营管理信息化功能指引
公立医院运营管理信息化功能指引的思维导图,为健全公立医院运营管理体系,提高运营管理科学化、规 范化、精细化、信息化水平,推动实现公立医院高质量发展, 本指引提出了公立医院运营管理信息化建设应用框架及功能设 计要求, 旨在引导各级各类公立医院运营管理信息化应用建设。
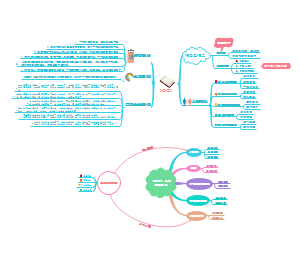
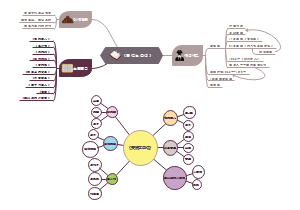
- 国家卫健委职能机构介绍
国家卫健委职能机构介绍的思维导图,分办公厅、人事司、规划发展与信息化司、财务司、法规司、体制改革司、医政司、基层卫生健康司、医疗应急司、科技教育司... ...
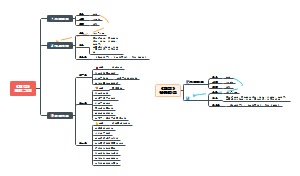
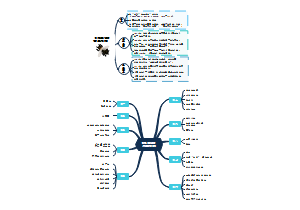
- 机器人技术体系汇总
前沿技术:与材料、化学、物理、生命科学、脑科学等交叉融合,产生颠覆性原创成果,与人工智能、大数据、云计算、5G、工业物联网等新技术的融合,产生率先突破的新技术。
ISO13485-2016医疗器械质量管理体系要求
社区模板帮助中心,点此进入>>
- 公立医院运营管理信息化功能指引
公立医院运营管理信息化功能指引的思维导图,为健全公立医院运营管理体系,提高运营管理科学化、规 范化、精细化、信息化水平,推动实现公立医院高质量发展, 本指引提出了公立医院运营管理信息化建设应用框架及功能设 计要求, 旨在引导各级各类公立医院运营管理信息化应用建设。
- 国家卫健委职能机构介绍
国家卫健委职能机构介绍的思维导图,分办公厅、人事司、规划发展与信息化司、财务司、法规司、体制改革司、医政司、基层卫生健康司、医疗应急司、科技教育司... ...
- 机器人技术体系汇总
前沿技术:与材料、化学、物理、生命科学、脑科学等交叉融合,产生颠覆性原创成果,与人工智能、大数据、云计算、5G、工业物联网等新技术的融合,产生率先突破的新技术。
- 相似推荐
- 大纲