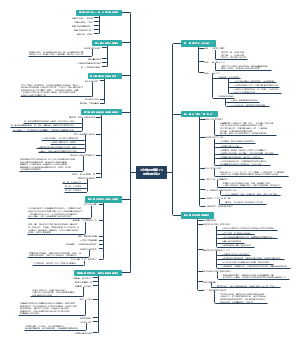
导图社区 基因治疗申报资料总体要求
基因治疗申报资料总体要求
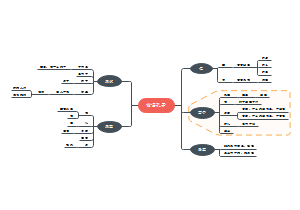
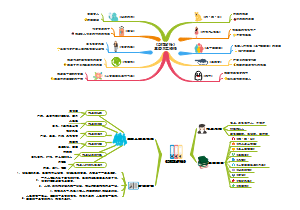
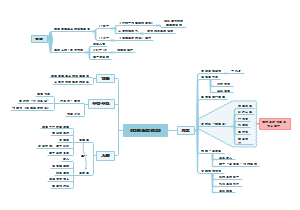
这是一篇关于申报资料总体要求的思维导图,主要内容包括:参考,物料管理,生产工艺,质量控制,稳定性。将申报资料总体要求涉及的各个方面进行了系统梳理,有助于相关人员全面了解和掌握申报资料的具体要求和内容。
编辑于2025-05-09 17:14:07- 质量控制
- 生产工艺
- 物料管理
他的近期作品
查看更多>>
- 先进治疗药品的范围、归类和释义
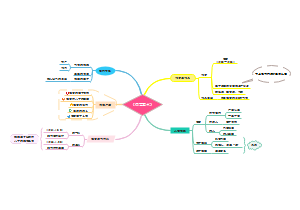
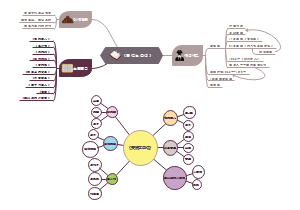
这是一篇关于ATMPS的思维导图,主要内容包括:前言,归类和释义,划分原则,有助于深入理解先进治疗药品的范畴和特性。
- 基因治疗申报资料总体要求
这是一篇关于申报资料总体要求的思维导图,主要内容包括:参考,物料管理,生产工艺,质量控制,稳定性。将申报资料总体要求涉及的各个方面进行了系统梳理,有助于相关人员全面了解和掌握申报资料的具体要求和内容。
- 腺相关病毒载体类体内基因治疗产品临床试验申请药学研究与评价技
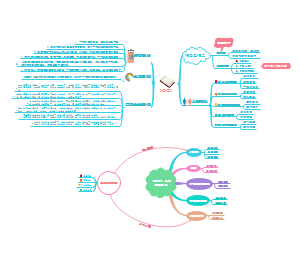
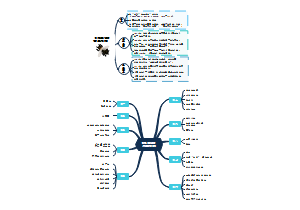
这是一篇关于腺相关病毒载体类体内基因治疗产品临床试验申请药学研究与评价技的思维导图,主要内容包括:目的,范围,一般原则,生产用材料,生产工艺,质量研究与质量标准,稳定性研究,包装与密封容器系统。
基因治疗申报资料总体要求
社区模板帮助中心,点此进入>>
他的近期作品
查看更多>>
- 先进治疗药品的范围、归类和释义
这是一篇关于ATMPS的思维导图,主要内容包括:前言,归类和释义,划分原则,有助于深入理解先进治疗药品的范畴和特性。
- 基因治疗申报资料总体要求
这是一篇关于申报资料总体要求的思维导图,主要内容包括:参考,物料管理,生产工艺,质量控制,稳定性。将申报资料总体要求涉及的各个方面进行了系统梳理,有助于相关人员全面了解和掌握申报资料的具体要求和内容。
- 腺相关病毒载体类体内基因治疗产品临床试验申请药学研究与评价技
这是一篇关于腺相关病毒载体类体内基因治疗产品临床试验申请药学研究与评价技的思维导图,主要内容包括:目的,范围,一般原则,生产用材料,生产工艺,质量研究与质量标准,稳定性研究,包装与密封容器系统。
- 相似推荐
- 大纲