导图社区 被动语态
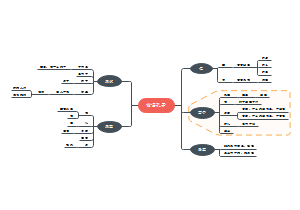
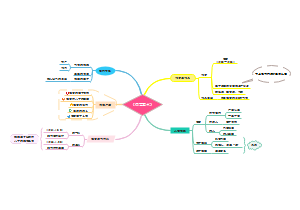
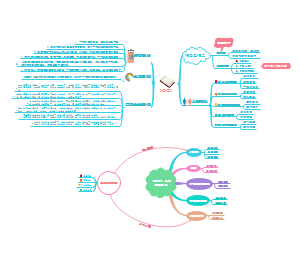
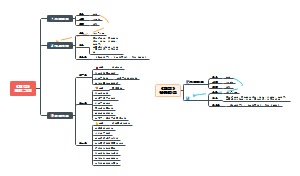
被动语态
不知道或不提及动作的执行者、强调动作的执行者、强调动作的承受者、动作的执行者是无生命的事物、常用于陈述客观事实,一般用在科技文章或者新闻报告中、部分动词短语/句型在习惯上只用于被动语态。
编辑于2023-01-09 12:33:09 广东- 初中英语语法
- 相似推荐
- 大纲
导图社区 被动语态
不知道或不提及动作的执行者、强调动作的执行者、强调动作的承受者、动作的执行者是无生命的事物、常用于陈述客观事实,一般用在科技文章或者新闻报告中、部分动词短语/句型在习惯上只用于被动语态。
编辑于2023-01-09 12:33:09 广东