导图社区 药品相关知识导图
药品相关知识导图
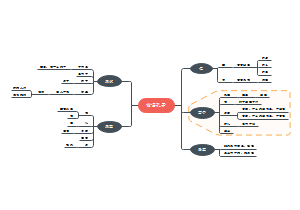
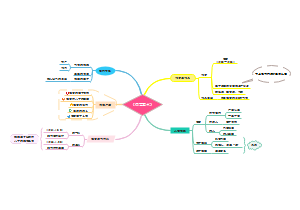
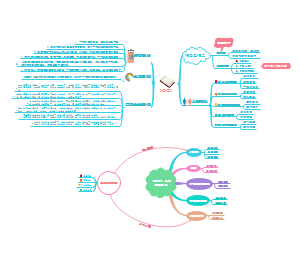
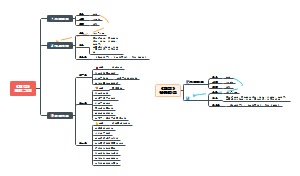
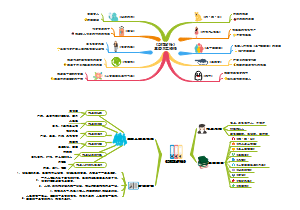
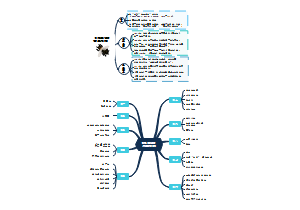
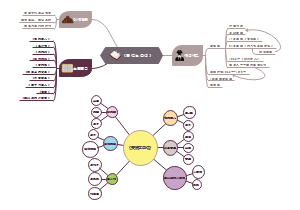
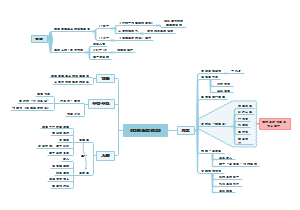
本图汇总的内容如下: 药品的申报与审批 药品注册其它规定和法律责任 药品经注册所取得的各种药品批准证明文件格式 药品知识产权概述 药品专利保护 药品专利的申请与授权 药品不良反应报告和监测的管理 药品不良反应报告的基本要求 药品不良反应报告与处置
编辑于2023-04-16 13:34:13- 相似推荐
- 大纲
导图社区 药品相关知识导图
本图汇总的内容如下: 药品的申报与审批 药品注册其它规定和法律责任 药品经注册所取得的各种药品批准证明文件格式 药品知识产权概述 药品专利保护 药品专利的申请与授权 药品不良反应报告和监测的管理 药品不良反应报告的基本要求 药品不良反应报告与处置
编辑于2023-04-16 13:34:13